Open Access | Review
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
Cardiovascular aging in patients with chronic kidney disease: pathogenesis and potential therapeutics
* Corresponding author: Mohamed Abdalbary
Mailing address: Lecturer of Nephrology, Mansoura Nephrology and Dialysis Unit, Mansoura University, 1 El Gomhouria St, Mansoura, Dakahlia Governorate 35516, Egypt.
Email: dr.mo7a.m@mans.edu.eg
* Corresponding author: Zahraa Alhoori
Mailing address: Medical Student, Mansoura Medical School, Mansoura University, 1 Tubli, Capital Governorate, Bahrain, Egypt.
Email: zahooralhoori@gmail.com
* Corresponding author: Safa Alkhayyat
Mailing address: Medical Student, Mansoura Medical School, Mansoura University, 1 Sitra, Capital Governorate, Bahrain, Egypt.
Email: safaa.alkhayyat@gmail.com
This article belongs to the Special Issue: Aging and Cardiovascular Diseases
Received: 01 August 2023 / Revised: 30 August 2023 / Accepted: 11 September 2023 / Published: 28 September 2023
DOI: 10.31491/APT.2023.09.118
Abstract
Patients with chronic kidney dysfunction have an elevated risk for various cardiovascular diseases. Even in the early stages of chronic kidney disease (CKD), the prevalence of cardiovascular events and mortality is extremely high if compared with the age-matched general population. With worsening of kidney function, this risk is growing intensely. There are many traditional and non-traditional risk factors that can lead to cardiovascular disease in CKD. Cardiovascular disease, rather than renal failure per se, is the major cause of mortality in CKD. The increase in calcification promoters and the decrease in inhibitors lead to the development of vascular calcification in the early stages of CKD. In this regard, CKD mimics cardiovascular system aging with a premature onset and an accelerated progression. Various non-pharmacological and pharmacological interventions have been studied to retard premature cardiovascular aging in CKD. In this review article, we summarized the pathogenesis, risk factors, and possible management strategies of cardiovascular disease in CKD.
Keywords
Cardiovascular, CKD, aging, vascular calcification, dialysis
Introduction
Chronic kidney disease (CKD) is characterized by abnormalities in kidney function or structure that persist for
more than three months. The severity of CKD is determined by the level of glomerular filtration rate (GFR) and
albuminuria [1]. Patients with advanced stages of CKD face a greater risk of cardiovascular events and death [2].
The incidence of CKD is estimated to be 13.4% of the world population, and it is progressively recognized as
a major public health issue that burdens societies and healthcare systems with significant medical and financial
costs [3, 4]. CKD can be described as a clinical model of premature aging. The aging process can be either pathogenic,
often known as premature aging or physiological. The slowly declining functional capacity leads to physiological
aging [5, 6]. Contrarily, premature aging is characterized by an accelerated functional decline that causes
aging to occur earlier than anticipated given chronological age [7]. Cardiovascular disease (CVD), persistent uremic
inflammation, osteoporosis, muscular atrophy and frailty are all characteristics of CKD.
CKD is associated with CVD, such as heart failure, arrhythmias, ischemic heart disease, and cardiac death. Patients
with advanced CKD stages demonstrate a noticeably augmented risk. The occurrence of cardiovascular events
is already higher in patients with mild kidney dysfunction compared to the general population. Cardiovascular disease—rather than kidney disease—is the major cause of
death in CKD. Long-lasting proinflammatory conditions induced by kidney disease enhance arterial calcification
and cardiac remodeling [8-10].
Vascular calcification (VC) is a sign of aging and a reliable predictor of cardiovascular morbidity and mortality
in the population with CKD. There is evidence that VC is predominant even in early CKD stages [11]. VC was once
thought to be a passive process, but it is now understood that VC is an invertible and highly controlled pathological
process and that the response to circulating calcification inhibitors, genetic factors, and hormones involves numerous
cellular signaling channels [12]. VC, which is a cellbased
process largely driven by vascular smooth muscle cells (VSMCs), mediates the accelerated early vascular
aging (EVA) [13]. Patients with CKD die prematurely
due to CVD even before many of them develop end-stage kidney disease (ESKD) [14]. In this review article, risk
factors, pathophysiology, and management of CVD in patients with CKD are disscussed.
Pathophysiology of premature vascular aging
There are traditional and non-traditional risk factors for premature vascular aging and calcification in CKD. Traditional risk factors for CKD include diabetes mellitus, dyslipidemia, hypertension, and obesity. On the other hand, non-traditional factors, include vascular calcification, phosphate imbalance, inflammation, oxidative stress, and cellular senescence. Figure 1 illustrates the non-traditional and traditional risk factors for cardiovascular aging in CKD.
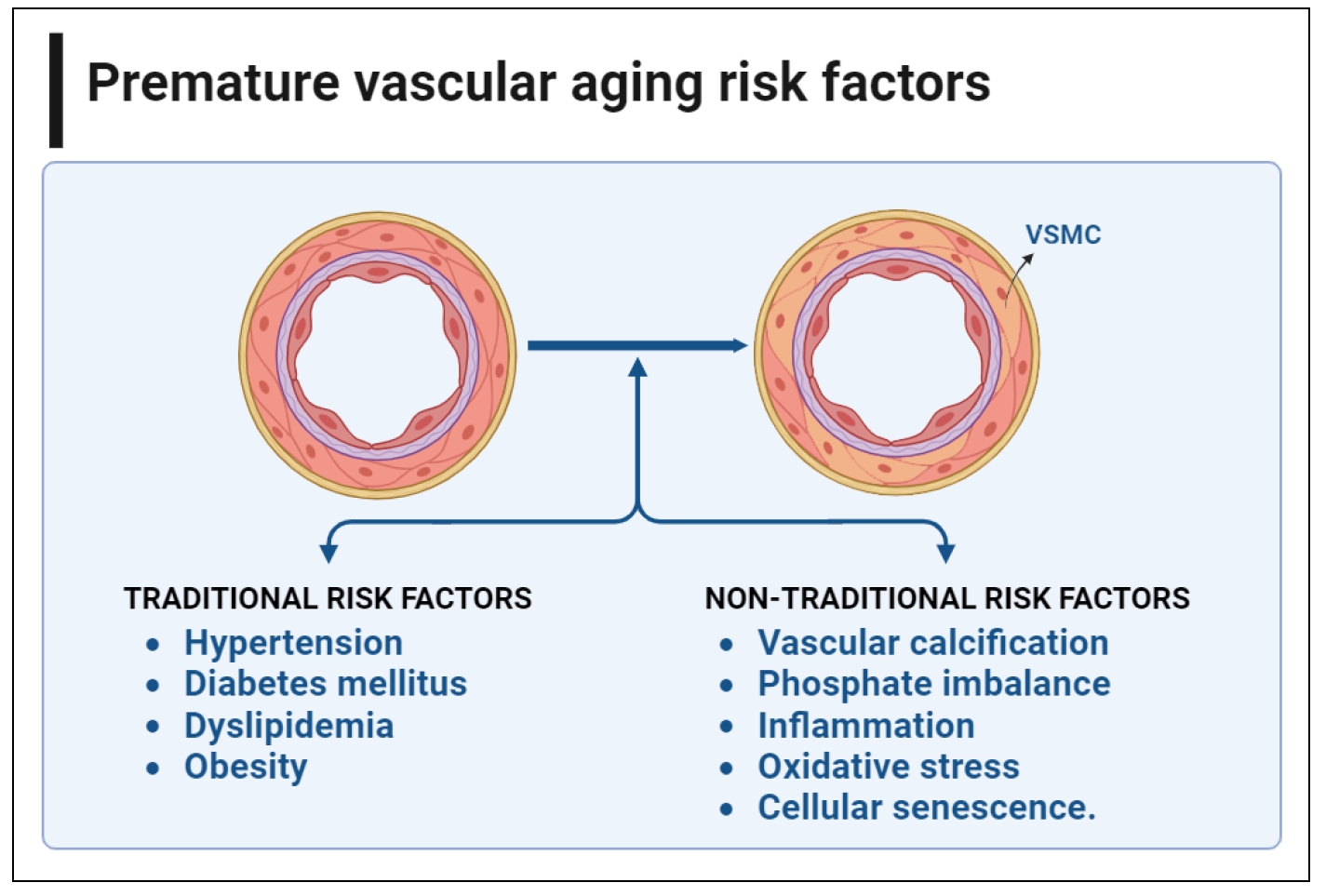
Figure 1. Non-traditional and traditional risk factorsfor cardiovascular aging in CKD. There are many traditional and nontraditional risk factors that promote vascular calcification and premature cardiovascular aging in CKD. Diabetes mellitus, dyslipidemia, hypertension, and obesity are among the most common traditional risk factors in patients with CKD. On the other hand, non-traditional factors, include vascular calcification, phosphate imbalance, inflammation, oxidative stress, and cellular senescence. This figure was created using BioRender.com.
Non-traditional risk factor
(1) Vascular calcification
VC is highly prevalent in patients with CKD and is closely associated with cardiovascular (CV) morbidity and mortality [15]. Vascular calcification can occur in tunica intima and/or tunica media. Calcification of the intimal layer forms atherosclerotic plaques and patchy crystals as a result of lipid and cholesterol deposition. It has been linked to smoking, dyslipidemia, and hypertension. In contrast, medial calcification usually occurs in the absence of lipid and cholesterol deposits and results in a sheet-like calcification and concentric thickening. Even in its early stages, patients with CKD are more likely to have medial calcification. It leads to decreased vascular compliance, causing increased arterial stiffness, resulting in impaired cardiac perfusion and progression of CVD. Medial calcification leads to an early vascular aging process (senescence) in patients with CKD. This premature aging is accompanied by chronic inflammation, continuous oxidative stress, DNA mutilation, and an imbalance of pro- and antiaging factors [16, 17].
There is accumulating evidence that VC is a cell-mediated pathological process that resembles the physiological bone formation by vascular smooth muscle cells (VSMCs) [18]. VSMCs are derived from the mesenchymal origin and under stress they can undergo osteogenic differentiation into another mesenchymal-derived cell type. VSMCs are present in the medial layer of vessels and play a fundamental role in regulating arterial tone and maintaining the vascular wall integrity [19].
In the patient with CKD, several factors can trigger calcification, including hypercalcemia, hyperphosphatemia, elevated levels of parathyroid hormone (PTH), inflammatory cytokines, oxidative stress, uremic toxins, advanced glycation end products [20]. Under normal circumstances, blood vessels are protected from excessive levels of serum calcium and phosphorus by various active inhibitors that prevent abnormal mineral accumulation in soft tissues. These inhibitors include pyrophosphate, adenosine, matrix Gla protein, osteopontin, fetuin-A, osteoprotegerin (OPG), and bone morphogenetic protein 2 (BMP-2) [21-24]. The increase in calcification inducers and the decrease in active inhibitors may explain the exceptionally high incidence of VC in CKD [25-27]. The uremic environment of CKD also encourages DNA damage, a major factor in cellular senescence, and stimulates osteogenic pathways in VSMCs, leading to the progression of VC [28]. There is increasing evidence that VC starts early and is predominant even in patients with mild renal impairment [11]. Figure 2 shows the frequently studied calcification promoters and inhibitors.
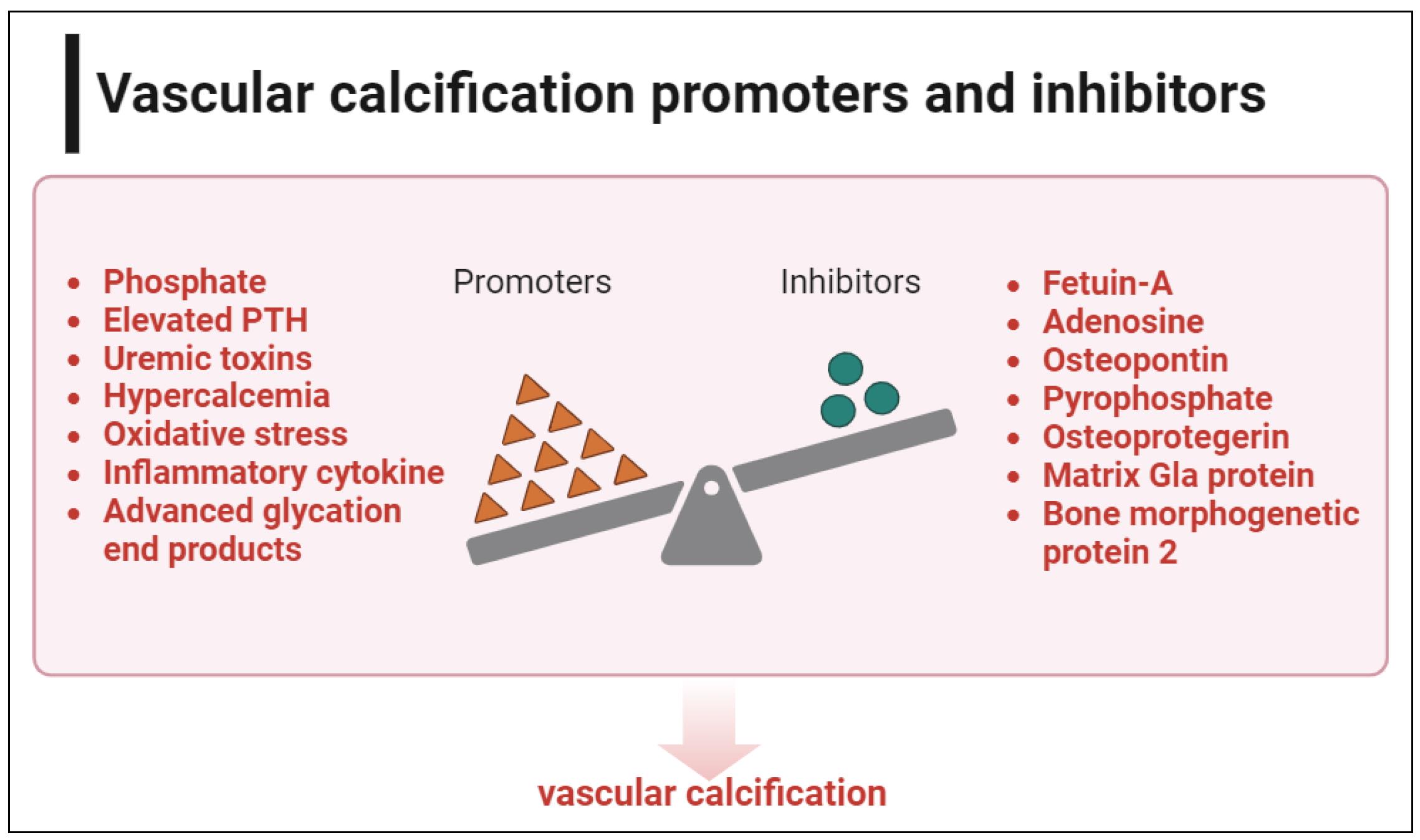
Figure 2. Vascular calcification promoters and inhibitors. In CKD, there is an imbalance between calcification promoters and inhibitors, leading to vascular calcification and premature cardiovascular aging. This figure was created using BioRender.com.
(2) Phosphate imbalance
Phosphate (Pi) levels are mainly maintained by the actions of three main players: the parathyroid hormone (PTH) and 1,25-dihydroxyvitamin D (Vit D), as well as fibroblast growth factor 23 (FGF-23) and its co-receptor, Klotho [29]. PTH and Vit D, the two main hormones, have antagonistic effects: PTH reduces the reabsorption of Pi in the kidney, whereas Vit D promotes this reabsorption and enhances intestinal absorption [30].
In CKD, Pi absorption and excretion are impaired, resulting in elevated Pi levels. FGF-23 and PTH are increased to keep Pi within the normal range by inducing hyperphosphaturia, but as the disease progresses, these systems are unable to maintain proper homeostasis, resulting in hyperphosphatemia [31]. Hyperphosphatemia is a key driver of VSMC differentiation into osteoblast-like cells [32]. Therefore, a wealth of data has shown that hyperphosphatemia negatively affects the cardiovascular system in CKD patients. High phosphate levels were linked to heart failure and an enlarged left ventricular mass even in the general population. However, a systematic review 2022 of 7 randomized clinical trials found no evidence of a reduction in cardiovascular risk in non-dialysis CKD patients with phosphate-lowering treatment [33]. Elevated FGF23 may induce cardiac damage and increase left ventricular hypertrophy (LVH) [34]. Additionally, epidemiologic studies have shown that FGF23 is linked to a higher risk of negative cardiovascular outcomes, including heart failure [35]. Low expression of Klotho, a cofactor of FGF receptors that has been identified as an anti-aging hormone, may play a role in this association. The precise molecular relationship between high FGF23 and CVD is still unknown. Compared to healthy people, soluble Klotho expression is lower in CKD patients, and they have a premature CV aging [36].
When the concentration of calcium and phosphate ions rises above the blood saturation level, amorphous calcium phosphate precipitates. This precipitate is then quickly absorbed by the serum protein fetuin-A to form calcium calciprotein monomers (CPMs), which then spontaneously aggregate to form primary calciprotein particles (CPPs). Secondary CPPs are created when primary CPPs aggregate and undergo a transition phase from the amorphous to the crystalline state of the calcium phosphate form. In cultured VSMC, secondary CPPs cause calcification, which is followed by inflammatory reactions [37].
(3) Oxidative stress
Excessive oxidative stress (OS) has been linked to the pathogenesis of VC [38]. Endoplasmic reticulum (ER) stress, which can be activated by OS, leads to VSMC differentiation into osteoblast-like cells. ER stress boosted XBP-1 expression, which has been demonstrated to bind to the Runx2 promoter, induce VSMC differentiation, and accelerate VSMC calcification [39]. In VSMCs and calcified aortas from experimental models, investigations discovered an increase in ER stress protein-activating transcription factor 4 (ATF4). Reduced ER stress, apoptosis, and VSMC calcification were seen with ATF4 RNA knockdown [40]. Simvastatin and ezetimibe may reduce ER stress and slow down VC in patients with renal dysfunction who had high OS [41].
(4) Inflammation
Clinical and epidemiologic studies have revealed a strong correlation between the risk of CV events and markers of inflammation in patients with CKD [42]. Traditional cardiovascular risk factors, such as HTN and hyperlipidemia, are linked to the inflammatory process in patients with CKD [43]. Moreover, several factors contribute to inflammation in CKD, including post-translational alteration of lipoproteins, infection, uremia, oxidative stress, insulin resistance, and accumulation of pro-inflammatory cytokines due to poor renal clearance [44]. Additionally, severe intestinal edema from CKD can cause overhydration, which can lead to bacterial or endotoxin translocation and systemic inflammation [45]. Indoxyl sulfate (IS) and pcresyl sulfate, two protein-bound uremic toxins that are not eliminated by conventional dialysis, promote inflammation and OS, leading to damage to vascular endothelial cell injury [46]. C reactive protein (CRP) and cytokines such as IL-6 and TNF-a levels in the plasma can be used to identify low-grade inflammation. In a long-term analysis, CRP assessed at baseline in the Modification of Diet in Renal Disease (MDRD) research was a reliable indicator of mortality from all causes and CVD [47-49]. In dialysis patients, the lower the CRP level, the lower the risk of mortality [50].
(5) Cellular senescence
Cellular senescence may play a crucial role in EVA and VSMC osteogenesis and calcification in CKD [51]. The accumulation and persistence of DNA damage is the primary factor causing cellular senescence. Senescent cells exhibit several pro-inflammatory and pro-fibrotic alterations in gene expression and cell metabolism, while losing their ability to divide but maintaining their metabolic activity. The senescence-associated secretory phenotype (SASP) is the name given to this novel trait. Growth factors, cytokines, proteases, and chemokines are more abundantly expressed and secreted in SASPs [52]. After an acute kidney injury, SASPs can help with tissue regeneration; however, long-term exposure to SASPs might promote sterile inflammation and speed up the development of CKD by encouraging renal fibrosis [53, 54]. Senescence and immune system dysfunction are two terms that are jointly referred to as immunosenescence [55]. Because immunosenescence is linked to low-grade sterile inflammation and diminished cellular defenses against infections and vaccinations, it is considered as hazardous [56]. BMP-2 and OPG, which are essential molecules in modulating calcification processes, were found to be secreted by aging VSMCs and may have activated osteogenic differentiation. This suggests a direct relationship between senescence and VC [57].
Traditional risk factor
In addition to the non-traditional risk factors, patients with CKD have many traditional risk factors which predispose to early vascular aging among these patients.
Diabetes mellitus (DM) and hypertension (HTN) are the two main causes of CKD worldwide [58] and are also major risk factors for CVD progression.
The kidney plays a significant role in regulating blood pressure, and HTN can predict the presence of underlying kidney disease. Inadequately managed hypertension can lead to a rapid decline in renal function, eventually resulting in ESKD. This can lead to a vicious circle [59]. CKD leads to the development of HTN by various causes, including sympathetic nervous system, sodium retention, and activation of the renin-angiotensin-aldosterone system (RAAS) [60-63]. HTN can both cause CKD and serve as a clinical indicator of the disease. According to the USRDS 2020, 72% of patients with CKD have hypertension [64].
There is strong evidence of the link between CVD and hypertension in patients with CKD. Patients with CKD who have hypertension have a 68% higher chance of developing CVD [65]. The link between hypertension and CVD in patients with CKD has been explained by several different mechanisms, including changes in the RAAS, oxidative stress, inflammation, and endothelial dysfunction [66]. The RAAS is known to be a noteworthy pathogenic component in VSMC proliferation, differentiation, and is likely to contribute to VC [67]. To lower the risk of CVD, the American Heart Association advises vigorous blood pressure control in patients with CKD [68].
Compared to non-diabetic patients, people with diabetes had more calcification. These patients had higher levels of osteopontin, type I collagen, and alkaline phosphatase in the medial layer of the arteries, which are bone matrix proteins [69]. It has been hypothesized that the advanced glycation end products (AGE) and their receptors for AGE (RAGE) facilitate the phenotypic transformation of VSMCs into osteoblast-like cells and trigger diabetesrelated VC [70, 71].
Obesity is a major precursor to diabetes and HTN. Moreover, it raises the risk of CKD and CVD [72, 73]. Obesity can have a direct impact on the heart, both pathologically and hemodynamically, by increasing myocardial fibrosis and volume overload [74]. In addition, obesity raises the risk of CVD through augmenting renal hyperfiltration and low-grade systemic inflammation [75]. Adipokines are a type of cytokine produced by cells such as adipocytes, macrophages, and lymphocytes, primarily in white adipose tissue. Depending on their relationship to the body’s inflammatory response, adipokines can have either anti-inflammatory or pro-inflammatory effects. Most adipokines are pro-inflammatory, including IL-6, TNF-α, A-FABP, PAI-1, resistin, leptin, and MCP-1. These proinflammatory adipokines are associated with metabolic and vascular complications related to obesity. In obesity, an increase in white adipose tissue from visceral deposits causes a shift toward pro-inflammatory molecules that can activate mechanisms leading to vascular calcification and arterial stiffness [76].
Dyslipidemia promotes the development and progression of VC by several pathways, one of which may be linked to cellular senescence. Modified low-density lipoprotein (LDL), triglyceride-rich lipoproteins (TRLs), and dysfunctional high-density lipoprotein (HDL) are the three primary types of pro-atherogenic lipids and lipoproteins. They can cause the senescence of several types of VC cells, such as endothelial cells (ECs), VSMCs, and adipose-derived mesenchymal stem cells (AMSCs). These senescent cells have a slower replication rate, more inflammation, and reactive oxygen species (ROS), all of which contribute to the development and progression of VC. Senescent ECs reduce endothelial integrity and permeability, allowing oxidized LDL to be retained and finally leading to VC [77].
Non-pharmacological interventions of cardiovascular disease in CKD
Non-pharmacological interventions are often overlooked; however, they are proven to be effective in slowing the progression of cardiovascular aging, generally without side effects. Advising patients for quitting smoking, regular muscle activity, dietary salt reduction, and weight loss are useful therapies at all CKD stages 8. There is a mutual association between CKD and aging. Elderly people with ESKD should be treated using a multifaceted treatment strategy that includes active rehabilitation as well [78]. Figure 3 illustrates the possible non-pharmacological interventions in patients with CKD.
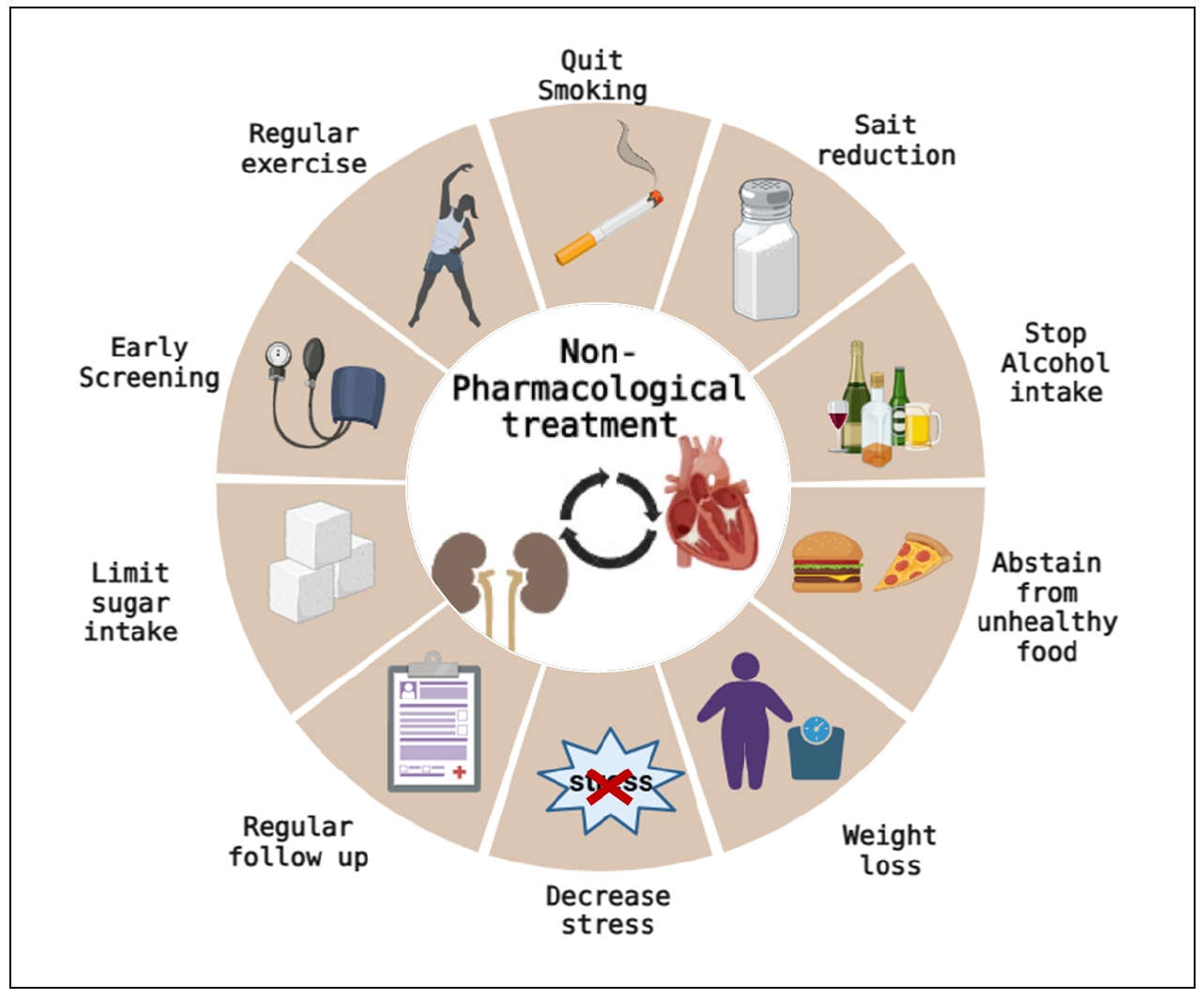
Figure 3. Nonpharmacological interventions of cardiovascular disease management in CKD. Non-pharmacological interventions are often overlooked; however, they can retard the progression of cardiovascular aging in CKD if properly advised and monitored. Advising patients for quitting smoking, regular exercise, salt reduction, and weight loss is beneficial at all CKD stages. Early screening and regular close follow-up can also help in the early management of cardiovascular disease. This figure was created using BioRender.com.
Pharmacological interventions of cardiovascular disease in CKD
Controlling DM and HTN are cornerstones of lowering cardiovascular risk in general population and in patients with CKD. Therefore, current recommendations call for strict control of blood pressure in patients with diabetic or non-diabetic CKD with RAAS blockers as the first-line medications [79].
Using SGLT2 inhibitors or GLP-1 receptor agonists, patients with type 2 diabetes have demonstrated a significant decrease in cardiovascular events. Guidelines therefore recommend using these medications to treat individuals with CKD and those without CKD who have CVD or multiple cardiovascular risk factors [80].
There is a shortage of data available to support management plans for cardiovascular risk in patients with CKD. Many approved and off-label drugs have been studied to decrease the vascular calcification in CKD.
Statin and aspirin
Dyslipidemia frequently occurs in CKD patients. The KDOQI advises that all adult patients with diabetic CKD and hypercholesterolemic non-diabetic CKD patients to receive treatment with a reductase inhibitor, or statin, to decrease LDL cholesterol. Statins help lessen a variety of cardiovascular complications brought on by atherosclerosis. According to recommendations, statins are advised for all CKD patients over 50 years of age and for people between the ages of 18 and 49 who are at high risk for atherosclerotic cardiovascular disease (CVD) [81].
The severity of CKD appears to have an impact on how well lipid-lowering therapies reduce CV risk in people with CKD. In patients with advanced CKD who had no prior history of myocardial infarction or coronary revascularization, the SHARP study discovered a significant relative decrease in the primary end point of cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, or coronary revascularization after using statins and ezetimibe [82].
In contrast, neither the 4D nor the AURORA investigations demonstrated a meaningful decrease in CVD in ESKD patients taking HD when compared to placebo [83]. According to these data, the cardiovascular benefits of lipid-lowering treatments are reduced with significant reduction of glomerular filtration rates and are only minimally effective in people with ESKD who are receiving hemodialysis [84].
Antiplatelet medications are well established to lower cardiovascular risk in people without CKD who have coronary artery disease, but the prognostic advantage is less obvious in CKD patients. Additionally, these medications raise the risk of bleeding incidents in CKD patients, thus may outweigh any potential advantages [85].
Phosphate binders
When dietary restriction is insufficient, patients with advanced CKD and hyperphosphatemia frequently need to be treated with phosphate binders. Since phosphate and the rise in FGF-23 and PTH that occurs along with it have all been linked to VC, lowering or keeping stable phosphate levels close to normal may be associated with improved overall CV system [85].
Using either calcium-containing (acetate, carbonate) or calcium-free (sevelamer, lanthanum, iron compounds, magnesium) binders, serum phosphate can be reduced to normal levels [86]. As a result of their ability to significantly lower dietary phosphate absorption, phosphate binders are the cornerstone of the therapy of patients with CKD and hyperphosphatemia [87, 88]. Non-calciumbased phosphate binders are generally preferable due to the possible risk of increased VC with calcium-based binders.
Phosphate binders successfully reduce urine phosphate excretion in studies done on healthy volunteers while maintaining serum phosphate levels within the usual range [89]. Moreover, phosphate binders—but not a placebo—reduce 24-hour urine phosphate in normophosphatemic patients with CKD stages 3–4 [90, 91]. Calcium-based binders did not decrease urinary phosphorus, possibly because calcium only serves as a secondary stimulator of the synthesis of FGF23 [92].
Calcimimetic
Calcimimetics can activate the parathyroid calcium-sensing receptor (CaSR), making parathyroid cells more sensitive to extracellular calcium. This inhibits the release of PTH and lowers serum calcium [93]. Patients with ESKD can be effectively treated for secondary hyperparathyroidism and by targeting CaSR, which is found in a variety of organs, but mainly in the parathyroid glands [94]. There is an evidence that VSMCs may include CaSR, as calcimimetics may directly influence the calcification process in these cells [95].
It appears that calcimimetics may slow down VC progression and decrease cardiovascular risk [96]. In one trial evaluating cinacalcet’s impact on cardiovascular morbidity and mortality, participants receiving cinacalcet saw significantly lower hospitalization rates and a tendency towards lower mortality [97]. Etelcalcetide, an intravenous calcimimetic that acts at a different location on the CaSR, outperformed cinacalcet on biochemical endpoints and was highly effective in reducing PTH and FGF-23. Although neither VC nor clinical outcomes have not been studied in relation to etelcalcetide effects [86].
Vitamin D
Vitamin D deficiency may have a major negative influence on CV risk. Vitamin D receptor activation has been associated to better blood pressure control and prevention of diabetic nephropathy [98]. On the other hand, natural calcitriol, a non-selective activator of vitamin D receptors, raises calcium and phosphate levels which would exacerbate the CV risk in CKD. Recent research revealed that paricalcitol, a selective VDRA, may have ameliorative effects on CV disease. Its potential benefit for diabetic nephropathy, cardiac illness, hypertension, and VC may pave the way for novel pathways in the treatment of CVD in patients with CKD [98].
PTH could be regulated in advanced CKD by active vitamin D. Retrospective studies have also revealed lower cardiovascular mortality in dialysis patients getting active vitamin D supplements [99]. Despite the limited number of clinical trials supporting the use of either native or active vitamin D analogues to stop the progression of VC, low doses of vitamin D or vitamin D analogues could be taken to prevent extremely high parathyroid hormone concentrations. On the other hand, low parathyroid hormone concentrations (oversuppression) are a noticeable side effect of overzealous use of calcium and vitamin D [8, 100, 101]. Repleting vitamin D deficiency with nutritional vitamin D, in addition to controlling PTH in patients with advanced CKD and secondary hyperparathyroidism with non-high calcium and phosphorus, could be beneficial for CVD management in patients with CKD.
Vitamin K
The protein matrix GIa protein (MGP), which depends on vitamin K for synthesis, has an inhibitory role in VC as it prevents the development of calcium crystals [102, 103]. To gain its anti-calcification capacity, vitamin K must decarboxylate MGP. Vitamin K antagonists use, vitamin K insufficiency and, as a result, decreased uncarboxylated MGP level have been associated with VC [94, 104].
Schurgers et al. demonstrated in animal models that undercarboxylation of MGP—caused by 6 weeks of therapy with the vitamin K antagonist warfarin—was related to accelerated VC [105]. Compared to rats receiving vitamin K supplements, the warfarin group showed quick VC, high atherogenic status, and notably higher levels of circulating undercarboxylated MGP, whereas high doses of vitamin K led to a 37% regression of VC status. This was the first in vivo study to demonstrate that vitamin K treatment may be able to prevent and even reverse vascular calcification [106].
There are no recommendations for the use of vitamin K supplements in patients with CKD. Of note, their use has not been associated with toxicity or serious side effects in any interventional research to yet. It could be a potentially safe supplement with probable benefit for CVD management in selected patients.
Magnesium
Recent studies have emphasized magnesium’s possible involvement in preventing vascular calcification [107, 108]. Few human clinical investigations have demonstrated that oral magnesium given to individuals with moderate to advanced CKD, in the form of a phosphate binder or as a supplement, may reduce VC progression or lowered the tendency for calcification [108].
Renal transplantation and renal replacement therapy
As renal function falls towards ESKD, important decisions regarding starting dialysis must be made. Regular or continuous dialysis treatments may be advantageous for CKD 5D patients with CHF [8, 100, 101]. Kidney transplantation may reverse uremia, which is a major trigger to development of VC in people with ESKD [81]. Preemptive kidney transplantation is the best option for patients with advanced CKD [101]. Patients with CKD who undergo kidney transplantation have some reduction in their cardiovascular risk [81].
Potential novel medications
The silent information regulator sirtuin 1 (SIRT1)
Through its control of fibrosis, apoptosis, and senescence, as well as oxidative stress, inflammation, VC, and aging process, SIRT1, a NAD+-dependent deacetylase, may play a protective role in CKD and its consequences on cardiovascular system. It could be a potential target for CVD management in CKD as it suppresses osteoblastic trans differentiation of VSMCs induced by hyperphosphatemia [109, 110].
SNF472: myo-inositol hexaphosphate
SNF472, a hexasodium salt of the active component, myo-inositol hexaphosphate (IP6) or phytate, has shown encouraging benefits in experimental trials. By adhering to hydroxyapatite crystal growth sites, SNF472 prevents the onset and development of calcification. This mechanism appears to be independent of the underlying cause of calcification and may offer a chance to block the final common pathway of VC [94].
Conclusions
In summary, CKD is a state of accelerated aging. Cardiovascular disease (CVD) is the leading cause of death in patients with CKD. Slowing the progression of CVD in CKD depends greatly on early detection and management of possible risk factors. CKD patients should maintain blood glucose and blood pressure control. Calcimimetics, non-calcium phosphate binders, and vitamin D have been used to control CKD mineral and bone disorders. Magnesium, vitamin K, and vitamin D could be potential therapies. New therapeutic agents and targets have been identified in recent years.
It is crucial to address the shortage of data from significant cardiovascular outcome studies in CKD with highrisk CVD. The most ideal strategy, till now, for advanced CKD may be kidney transplantation, which can improve ESKD-related cardiovascular outcomes.
Declarations
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
The authors report no conflicts of interest.
Ethical approval and informed consent statement
Not applicable.
Consent for publication
Not applicable.
References
1. Shori AB. Camel milk as a potential therapy for controlling diabetes and its complications: a review of in vivo studies. J Food Drug Anal, 2015, 23(4): 609-618. [Crossref]
2. Yamany A, Shehata H, Essameldin M, Ibrahim S. Screening of incidental kidney disease in normoglycemic, normotensive healthy adults. The Egyptian Journal of Internal Medicine, 2017, 29: 127-131.
3. Eckardt KU, Coresh J, Devuyst O, Johnson RJ, Köttgen A, Levey AS, et al. Evolving importance of kidney disease: from subspecialty to global health burden. Lancet, 2013, 382(9887): 158-169. [Crossref]
4. Hill NR, Fatoba ST, Oke JL, Hirst JA, O’Callaghan CA, Lasserson DS, et al. Global prevalence of chronic kidney disease - A systematic review and meta-analysis. PLoS One, 2016, 11(7): e0158765. [Crossref]
5. Gadecka A, Bielak-Zmijewska A. Slowing down aging: the role of nutrients and microbiota in modulation of the epigenome. Nutrients, 2019, 11(6). [Crossref]
6. Morales-Vives F, Dueñas JM. Predicting suicidal ideation in adolescent boys and girls: the role of psychological maturity, personality traits, depression and life satisfaction. Span J Psychol, 2018, 21: E10. [Crossref]
7. Hamczyk MR, Nevado RM, Barettino A, Fuster V, Andrés V. Biological versus chronological aging: JACC focus seminar. J Am Coll Cardiol, 2020, 75(8): 919-930. [Crossref]
8. Herzog CA, Asinger RW, Berger AK, Charytan DM, Díez J, Hart RG, et al. Cardiovascular disease in chronic kidney disease. A clinical update from kidney disease: improving global outcomes (KDIGO). Kidney Int, 2011, 80(6): 572-586. [Crossref]
9. Matsushita K, Ballew SH, Wang AY, Kalyesubula R, Schaeffner E, Agarwal R. Epidemiology and risk of cardiovascular disease in populations with chronic kidney disease. Nat Rev Nephrol, 2022, 18(11): 696-707. [Crossref]
10. Ortiz A, Wanner C, Gansevoort R. Chronic kidney disease as cardiovascular risk factor in routine clinical practice: a position statement by the Council of the European Renal Association. Clin Kidney J, 2023, 16(3): 403-407. [Crossref]
11. El-Husseini A, Abdalbary M, Lima F, Issa M, Ahmed MT, Winkler M, et al. Low Turnover renal osteodystrophy with abnormal bone quality and vascular calcification in patients with mild-to-moderate CKD. Kidney Int Rep, 2022, 7(5): 1016-1026. [Crossref]
12. Shioi A, Ikari Y. Plaque Calcification during atherosclerosis progression and regression. J Atheroscler Thromb, 2018, 25(4): 294-303. [Crossref]
13. Dai L, Qureshi AR, Witasp A, Lindholm B, Stenvinkel P. Early vascular ageing and cellular senescence in chronic kidney disease. Comput Struct Biotechnol J, 2019, 17: 721-729. [Crossref]
14. Suarez J, Cohen JB, Potluri V, Yang W, Kaplan DE, Serper M, et al. Racial disparities in nephrology consultation and disease progression among veterans with CKD: an observational cohort study. J Am Soc Nephrol, 2018, 29(10): 2563-2573. [Crossref]
15. Tonelli M, Karumanchi SA, Thadhani R. Epidemiology and mechanisms of uremia-related cardiovascular disease. Circulation, 2016, 133(5): 518-536. [Crossref]
16. Kakani E, Elyamny M, Ayach T, El-Husseini A. Pathogenesis and management of vascular calcification in CKD and dialysis patients. Semin Dial, 2019, 32(6): 553-561. [Crossref]
17. Shanahan CM, Cary NR, Salisbury JR, Proudfoot D, Weissberg PL, Edmonds ME. Medial localization of mineralization-regulating proteins in association with Mönckeberg’s sclerosis: evidence for smooth muscle cell-mediated vascular calcification. Circulation, 1999, 100(21): 2168-2176. [Crossref]
18. Smith ER. Vascular Calcification in uremia: new-age concepts about an old-age problem. Methods Mol Biol, 2016, 1397: 175-208. [Crossref]
19. Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res, 2012, 95(2): 156-164. [Crossref]
20. Paloian NJ, Giachelli CM. A current understanding of vascular calcification in CKD. Am J Physiol Renal Physiol, 2014, 307(8): F891-900. [Crossref]
21. Schafer C, Heiss A, Schwarz A, Westenfeld R, Ketteler M, Floege J, et al. The serum protein alpha 2-Heremans- Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest, 2003, 112(3): 357-366. [Crossref]
22. Bennett BJ, Scatena M, Kirk EA, Rattazzi M, Varon RM, Averill M, et al. Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older ApoE-/- mice. Arterioscler Thromb Vasc Biol, 2006, 26(9): 2117-2124. [Crossref]
23. McCabe KM, Booth SL, Fu X, Shobeiri N, Pang JJ, Adams MA, et al. Dietary vitamin K and therapeutic warfarin alter the susceptibility to vascular calcification in experimental chronic kidney disease. Kidney Int, 2013, 83(5): 835-844. [Crossref]
24. O’Neill WC, Lomashvili KA, Malluche HH, Faugere MC, Riser BL. Treatment with pyrophosphate inhibits uremic vascular calcification. Kidney Int, 2011, 79(5): 512-517. [Crossref]
25. Leskinen Y, Paana T, Saha H, Groundstroem K, Lehtimäki T, Kilpinen S, et al. Valvular calcification and its relationship to atherosclerosis in chronic kidney disease. J Heart Valve Dis, 2009, 18(4): 429-438.
26. Cannata-Andia JB, Roman-Garcia P, Hruska K. The connections between vascular calcification and bone health. Nephrol Dial Transplant, 2011, 26(11): 3429-3436. [Crossref]
27. Oliveira RBd, Okazaki H, Stinghen AEM, Drüeke TB, Massy ZA, Jorgetti V. Vascular calcification in chronic kidney disease: a review. Brazilian Journal of Nephrology, 2013, 35: 147-161.
28. Shanahan CM. Mechanisms of vascular calcification in CKD-evidence for premature ageing? Nat Rev Nephrol, 2013, 9(11): 661-670. [Crossref]
29. Toapanta Gaibor NG, Nava Pérez NC, Martínez Echevers Y, Montes Delgado R, Guerrero Riscos M. PTH levels and not serum phosphorus levels are a predictor of the progression of kidney disease in elderly patients with advanced chronic kidney disease. Nefrologia, 2017, 37(2): 149-157. [Crossref]
30. Berndt TJ, Schiavi S, Kumar R. “Phosphatonins” and the regulation of phosphorus homeostasis. Am J Physiol Renal Physiol, 2005, 289(6): F1170-1182. [Crossref]
31. Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab, 2006, 291(1): E38-49. [Crossref]
32. Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res, 2011, 109(6): 697-711. [Crossref]
33. Lioufas NM, Pascoe EM, Hawley CM, Elder GJ, Badve SV, Block GA, et al. Systematic review and meta-analyses of the effects of phosphate-lowering agents in nondialysis CKD. J Am Soc Nephrol, 2022, 33(1): 59-76. [Crossref]
34. Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest, 2011, 121(11): 4393-4408. [Crossref]
35. Gutiérrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med, 2008, 359(6): 584-592. [Crossref]
36. Koh N, Fujimori T, Nishiguchi S, Tamori A, Shiomi S, Nakatani T, et al. Severely reduced production of klotho in human chronic renal failure kidney. Biochem Biophys Res Commun, 2001, 280(4): 1015-1020. [Crossref]
37. Mizuno Y, Ishida T, Kugimiya F, Takai S, Nakayama Y, Yonemitsu K, et al. Deterioration of phosphate homeostasis is a trigger for cardiac afterload - clinical importance of fibroblast growth factor 23 for accelerated aging. Circ Rep, 2023, 5(3): 103-104. [Crossref]
38. Evrard S, Delanaye P, Kamel S, Cristol JP, Cavalier E. Vascular calcification: from pathophysiology to biomarkers. Clin Chim Acta, 2015, 438: 401-414. [Crossref]
39. Liberman M, Johnson RC, Handy DE, Loscalzo J, Leopold JA. Bone morphogenetic protein-2 activates NADPH oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification. Biochem Biophys Res Commun, 2011, 413(3): 436-441. [Crossref]
40. Duan XH, Chang JR, Zhang J, Zhang BH, Li YL, Teng X, et al. Activating transcription factor 4 is involved in endoplasmic reticulum stress-mediated apoptosis contributing to vascular calcification. Apoptosis, 2013, 18(9): 1132-1144. [Crossref]
41. Miyazaki-Anzai S, Masuda M, Demos-Davies KM, Keenan AL, Saunders SJ, Masuda R, et al. Endoplasmic reticulum stress effector CCAAT/enhancer-binding protein homologous protein (CHOP) regulates chronic kidney diseaseinduced vascular calcification. J Am Heart Assoc, 2014, 3(3): e000949. [Crossref]
42. Cottone S, Lorito MC, Riccobene R, Nardi E, Mulè G, Buscemi S, et al. Oxidative stress, inflammation and cardiovascular disease in chronic renal failure. J Nephrol, 2008, 21(2): 175-179.
43. Silverstein DM. Inflammation in chronic kidney disease: role in the progression of renal and cardiovascular disease. Pediatr Nephrol, 2009, 24(8): 1445-1452. [Crossref]
44. Zoccali C, Vanholder R, Massy ZA, Ortiz A, Sarafidis P, Dekker FW, et al. The systemic nature of CKD. Nat Rev Nephrol, 2017, 13(6): 344-358. [Crossref]
45. Pham PT, Pham PA, Pham PC, Parikh S, Danovitch G. Evaluation of adult kidney transplant candidates. Semin Dial, 2010, 23(6): 595-605. [Crossref]
46. Ramezani A, Raj DS. The gut microbiome, kidney disease, and targeted interventions. J Am Soc Nephrol, 2014, 25(4): 657-670. [Crossref]
47. Kumar V, Baruah K, Nguyen DV, Smagghe G, Vossen E, Bossier P. Phloroglucinol-mediated hsp70 production in crustaceans: protection against Vibrio parahaemolyticus in artemia franciscana and macrobrachium rosenbergii. Front Immunol, 2020, 11: 1860. [Crossref]
48. Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of diet in renal disease study group. Ann Intern Med, 1999, 130(6): 461-470. [Crossref]
49. Henze LA, Luong TTD, Boehme B, Masyout J, Schneider MP, Brachs S, et al. Impact of C-reactive protein on osteo-/chondrogenic transdifferentiation and calcification of vascular smooth muscle cells. Aging (Albany NY),2019, 11(15): 5445-5462. [Crossref]
50. Senjem ML, Gunter JL, Shiung MM, Petersen RC, Jack CR, Jr. Comparison of different methodological implementations of voxel-based morphometry in neurodegenerative disease. Neuroimage, 2005, 26(2): 600-608. [Crossref]
51. Dai L, Schurgers LJ, Shiels PG, Stenvinkel P. Early vascular ageing in chronic kidney disease: impact of inflammation, vitamin K, senescence and genomic damage. Nephrol Dial Transplant, 2020, 35(Suppl 2): ii31-ii37. [Crossref]
52. Karthik L, Kumar G, Keswani T, Bhattacharyya A, Chandar SS, Bhaskara Rao KV. Protease inhibitors from marine actinobacteria as a potential source for antimalarial compound. PLoS One, 2014, 9(3): e90972. [Crossref]
53. Sturmlechner I, Durik M, Sieben CJ, Baker DJ, van Deursen JM. Cellular senescence in renal ageing and disease. Nat Rev Nephrol, 2017, 13(2): 77-89. [Crossref]
54. Wang WJ, Cai GY, Chen XM. Cellular senescence, senescence-associated secretory phenotype, and chronic kidney disease. Oncotarget, 2017, 8(38): 64520-64533. [Crossref]
55. Santoro A, Bientinesi E, Monti D. Immunosenescence and inflammaging in the aging process: age-related diseases or longevity? Ageing Res Rev, 2021, 71: 101422. [Crossref]
56. Pawelec G. Age and immunity: What is “immunosenescence”? Exp Gerontol, 2018, 105: 4-9. [Crossref]
57. Liu Y, Drozdov I, Shroff R, Beltran LE, Shanahan CM. Prelamin a accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ Res, 2013, 112(10): e99-109. [Crossref]
58. Kanno Y, Into T, Lowenstein CJ, Matsushita K. Nitric oxide regulates vascular calcification by interfering with TGF- signalling. Cardiovasc Res, 2008, 77(1): 221-230. [Crossref]
59. Zaragoza C, López-Rivera E, García-Rama C, Saura M, Martínez-Ruíz A, Lizarbe TR, et al. Cbfa-1 mediates nitric oxide regulation of MMP-13 in osteoblasts. J Cell Sci, 2006, 119(Pt 9): 1896-1902. [Crossref]
60. Cao X, Li H, Tao H, Wu N, Yu L, Zhang D, et al. Metformin inhibits vascular calcification in female rat aortic smooth muscle cells via the AMPK-eNOS-NO pathway. Endocrinology, 2013, 154(10): 3680-3689. [Crossref]
61. Soskić SS, Dobutović BD, Sudar EM, Obradović MM, Nikolić DM, Djordjevic JD, et al. Regulation of inducible nitric oxide synthase (iNOS) and its potential role in insulin resistance, diabetes and heart failure. Open Cardiovasc Med J, 2011, 5: 153-163. [Crossref]
62. Chang XY, Cui L, Wang XZ, Zhang L, Zhu D, Zhou XR, et al. Quercetin attenuates vascular calcification through suppressed oxidative stress in adenine-induced chronic renal failure rats. Biomed Res Int, 2017, 2017: 5716204. [Crossref]
63. Gloria MAD, Mouro MG, Geraldini S, Higa EMS, Carvalho AB. Cbfa1 expression in vascular smooth muscle cells may be elevated by increased nitric oxide/iNOS. J Bras Nefrol, 2020, 42(3): 300-306. [Crossref]
64. Wei Q, Ren X, Jiang Y, Jin H, Liu N, Li J. Advanced glycation end products accelerate rat vascular calcification through RAGE/oxidative stress. BMC Cardiovasc Disord, 2013, 13: 13. [Crossref]
65. Xie X, Liu Y, Perkovic V, Li X, Ninomiya T, Hou W, et al. Renin-angiotensin system inhibitors and kidney and cardiovascular outcomes in patients with CKD: a bayesian network meta-analysis of randomized clinical trials. Am J Kidney Dis, 2016, 67(5): 728-741 [Crossref]
66. Phan O, Burnier M, Wuerzner G. Hypertension in chronic kidney disease - Role of arterial calcification and impact on treatment. Eur Cardiol, 2014, 9(2): 115-119. [Crossref]
67. Savoia C, Burger D, Nishigaki N, Montezano A, Touyz RM. Angiotensin II and the vascular phenotype in hypertension. Expert Rev Mol Med, 2011, 13: e11. [Crossref]
68. Whelton PK, Carey RM, Aronow WS, Casey DE, Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. Journal of the American College of Cardiology, 2018, 71(19): e127-e248.
69. Boström KI, Jumabay M, Matveyenko A, Nicholas SB, Yao Y. Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circ Res, 2011, 108(4): 446-457. [Crossref]
70. Chen NX, Duan D, O’Neill KD, Moe SM. High glucose increases the expression of Cbfa1 and BMP-2 and enhances the calcification of vascular smooth muscle cells. Nephrol Dial Transplant, 2006, 21(12): 3435-3442. [Crossref]
71. Chen NX, Duan D, O’Neill KD, Wolisi GO, Koczman JJ, Laclair R, et al. The mechanisms of uremic seruminduced expression of bone matrix proteins in bovine vascular smooth muscle cells. Kidney Int, 2006, 70(6): 1046-1053. [Crossref]
72. Chang AR, Grams ME, Ballew SH, Bilo H, Correa A, Evans M, et al. Adiposity and risk of decline in glomerular filtration rate: meta-analysis of individual participant data in a global consortium. BMJ, 2019, 364: k5301. [Crossref]
73. Wormser D, Kaptoge S, Di Angelantonio E, Wood AM, Pennells L, Thompson A, et al. Separate and combined associations of body-mass index and abdominal adiposity with cardiovascular disease: collaborative analysis of 58 prospective studies. Lancet, 2011, 377(9771): 1085-1095. [Crossref]
74. Powell-Wiley TM, Poirier P, Burke LE, Després JP, Gordon-Larsen P, Lavie CJ, et al. Obesity and cardiovascular disease: a scientific statement from the American heart association. Circulation, 2021, 143(21): e984-e1010. [Crossref]
75. Ellulu MS, Patimah I, Khaza’ai H, Rahmat A, Abed Y. Obesity and inflammation: the linking mechanism and the complications. Arch Med Sci, 2017, 13(4): 851-863. [Crossref]
76. Para I, Albu A, Porojan MD. Adipokines and arterial stiffness in obesity. Medicina (Kaunas), 2021, 57(7). [Crossref]
77. Xiang Q, Tian F, Xu J, Du X, Zhang S, Liu L. New insight into dyslipidemia-induced cellular senescence in atherosclerosis. Biol Rev Camb Philos Soc, 2022, 97(5): 1844-1867. [Crossref]
78. Kooman JP, van der Sande FM, Leunissen KM. Kidney disease and aging: A reciprocal relation. Exp Gerontol, 2017, 87(Pt B): 156-159. [Crossref]
79. Williams B, Mancia G, Spiering W, Rosei EA, Azizi M, Burnier M, et al. 2018 ESC/ESH guidelines for the management of arterial hypertension. Kardiologia Polska (Polish Heart Journal), 2019, 77(2): 71-159.
80. Jankowski J, Floege J, Fliser D, Böhm M, Marx N. Cardiovascular disease in chronic kidney disease: pathophysiological insights and therapeutic options. Circulation, 2021, 143(11): 1157-1172. [Crossref]
81. Mathew RO, Bangalore S, Lavelle MP, Pellikka PA, Sidhu MS, Boden WE, et al. Diagnosis and management of atherosclerotic cardiovascular disease in chronic kidney disease: a review. Kidney Int, 2017, 91(4): 797-807. [Crossref]
82. Sharp Collaborative G. Study of heart and renal protection (SHARP): randomized trial to assess the effects of lowering low-density lipoprotein cholesterol among 9,438 patients with chronic kidney disease. Am Heart J, 2010, 160(5): 785-794.e710. [Crossref]
83. Baigent C, Landray MJ, Reith C, Emberson J, Wheeler DC, Tomson C, et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (study of heart and renal protection): a randomised placebo-controlled trial. Lancet, 2011, 377(9784): 2181-2192. [Crossref]
84. Palmer SC, Craig JC, Navaneethan SD, Tonelli M, Pellegrini F, Strippoli GF. Benefits and harms of statin therapy for persons with chronic kidney disease: a systematic review and meta-analysis. Ann Intern Med, 2012, 157(4): 263-275. [Crossref]
85. Adeney KL, Siscovick DS, Ix JH, Seliger SL, Shlipak MG, Jenny NS, et al. Association of serum phosphate with vascular and valvular calcification in moderate CKD. J Am Soc Nephrol, 2009, 20(2): 381-387. [Crossref]
86. Nelson AJ, Raggi P, Wolf M, Gold AM, Chertow GM, Roe MT. Targeting vascular calcification in chronic kidney disease. JACC Basic Transl Sci, 2020, 5(4): 398-412. [Crossref]
87. Daugirdas JT, Finn WF, Emmett M, Chertow GM. The phosphate binder equivalent dose. Semin Dial, 2011, 24(1): 41-49. [Crossref]
88. Tonelli M, Pannu N, Manns B. Oral phosphate binders in patients with kidney failure. N Engl J Med, 2010, 362(14): 1312-1324. [Crossref]
89. Pennick M, Poole L, Dennis K, Smyth M. Lanthanum carbonate reduces urine phosphorus excretion: evidence of high-capacity phosphate binding. Ren Fail, 2012, 34(3): 263-270. [Crossref]
90. Oliveira RB, Cancela AL, Graciolli FG, Dos Reis LM, Draibe SA, Cuppari L, et al. Early control of PTH and FGF23 in normophosphatemic CKD patients: a new target in CKD-MBD therapy? Clin J Am Soc Nephrol, 2010, 5(2): 286-291. [Crossref]
91. Hill KM, Martin BR, Wastney ME, McCabe GP, Moe SM, Weaver CM, et al. Oral calcium carbonate affects calcium but not phosphorus balance in stage 3-4 chronic kidney disease. Kidney Int, 2013, 83(5): 959-966. [Crossref]
92. Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, et al. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res, 2000, 87(7): E10-17. [Crossref]
93. Neven E, D’Haese PC. Vascular calcification in chronic renal failure: what have we learned from animal studies? Circ Res, 2011, 108(2): 249-264. [Crossref]
94. Nelson AJ, Raggi P, Wolf M, Gold AM, Chertow GM, Roe MT. Targeting vascular calcification in chronic kidney disease. Basic to Translational Science, 2020, 5(4): 398-412.
95. Molostvov G, James S, Fletcher S, Bennett J, Lehnert H, Bland R, et al. Extracellular calcium-sensing receptor is functionally expressed in human artery. Am J Physiol Renal Physiol, 2007, 293(3): F946-955. [Crossref]
96. Joki N, Nikolov IG, Caudrillier A, Mentaverri R, Massy ZA, Drüeke TB. Effects of calcimimetic on vascular calcification and atherosclerosis in uremic mice. Bone, 2009, 45 Suppl 1: S30-34. [Crossref]
97. Cunningham J, Danese M, Olson K, Klassen P, Chertow GM. Effects of the calcimimetic cinacalcet HCl on cardiovascular disease, fracture, and health-related quality of life in secondary hyperparathyroidism. Kidney Int, 2005, 68(4): 1793-1800. [Crossref]
98. Cozzolino M, Stucchi A, Rizzo MA, Soldati L, Cusi D, Ciceri P, et al. Vitamin D receptor activation and prevention of arterial ageing. Nutr Metab Cardiovasc Dis, 2012, 22(7): 547-552. [Crossref]
99. Hou YC, Liu WC, Zheng CM, Zheng JQ, Yen TH, Lu KC. Role of Vitamin D in uremic vascular calcification. Biomed Res Int, 2017, 2017: 2803579. [Crossref]
100. Chou YH, Chen YM. Aging and renal disease: old questions for new challenges. Aging Dis, 2021, 12(2): 515-528. [Crossref]
101. Sharaf El Din UA, Salem MM, Abdulazim DO. Vascular calcification: when should we interfere in chronic kidney disease patients and how? World J Nephrol, 2016, 5(5): 398-417. [Crossref]
102. Boström K, Tsao D, Shen S, Wang Y, Demer LL. Matrix GLA protein modulates differentiation induced by bone morphogenetic protein-2 in C3H10T1/2 cells. J Biol Chem, 2001, 276(17): 14044-14052. [Crossref]
103. Jaminon AMG, Dai L, Qureshi AR, Evenepoel P, Ripsweden J, Söderberg M, et al. Matrix Gla protein is an independent predictor of both intimal and medial vascular calcification in chronic kidney disease. Sci Rep, 2020, 10(1): 6586. [Crossref]
104. Parker BD, Ix JH, Cranenburg EC, Vermeer C, Whooley MA, Schurgers LJ. Association of kidney function and uncarboxylated matrix Gla protein: data from the Heart and Soul Study. Nephrol Dial Transplant, 2009, 24(7): 2095-2101. [Crossref]
105. Roumeliotis S, Dounousi E, Salmas M, Eleftheriadis T, Liakopoulos V. Vascular calcification in chronic kidney disease: the role of vitamin K-dependent matrix Gla protein. Frontiers in medicine, 2020, 7: 154.
106. Roumeliotis S, Dounousi E, Salmas M, Eleftheriadis T, Liakopoulos V. Vascular calcification in chronic kidney disease: the role of Vitamin K- dependent matrix Gla Protein. Front Med (Lausanne), 2020, 7: 154. [Crossref]
107. Kircelli F, Peter ME, Sevinc Ok E, Celenk FG, Yilmaz M, Steppan S, et al. Magnesium reduces calcification in bovine vascular smooth muscle cells in a dose-dependent manner. Nephrol Dial Transplant, 2012, 27(2): 514-521. [Crossref]
108. Xu J, Bai Y, Jin J, Zhang J, Zhang S, Cui L, et al. Magnesium modulates the expression levels of calcification-associated factors to inhibit calcification in a time-dependent manner. Exp Ther Med, 2015, 9(3): 1028-1034. [Crossref]
109. Takemura A, Iijima K, Ota H, Son BK, Ito Y, Ogawa S, et al. Sirtuin 1 retards hyperphosphatemia-induced calcification of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol, 2011, 31(9): 2054-2062. [Crossref]
110. Yan J, Wang J, He JC, Zhong Y. Sirtuin 1 in chronic kidney disease and therapeutic potential of targeting Sirtuin 1. Front Endocrinol (Lausanne), 2022, 13: 917773. [Crossref]