Open Access | Review
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
Accelerated biological aging in type 2 diabetes mellitus: mechanistic, cellular, and systemic perspectives
* Corresponding author: Simran C. Yadav
Mailing address: Department of Pharmacology, JES’s SND College of Pharmacy, Babhulgaon (Yeola), India.
Email: simranyd2423@gmail.com
Received: 12 January 2026 / Revised: 06 February 2026 / Accepted: 27 February 2026 / Published: 31 March 2026
DOI: 10.31491/APT.2026.03.203
Abstract
Type 2 diabetes mellitus (T2DM), a common chronic metabolic disease that affects multiple organ systems, is still a major global public health concern. Long-term diabetes is linked to cardiovascular disease, renal impairment, neurotoxicity, musculoskeletal weakness, and progressive pancreatic β-cell loss in addition to disruptions in glucose metabolism. These issues frequently start earlier and worsen faster than one may anticipate given one’s age. According to recent research, T2DM may be considered a disorder of accelerated biological aging. Diabetes patients exhibit early activation of aging-related processes, including cellular senescence, oxidative stress, chronic low-grade inflammation, genomic instability, mitochondrial dysfunction, and epigenetic changes. Persistent hyperglycemia, insulin resistance, and metabolic stress intensify these pathways, which are similar to those seen during physiological aging. The main molecular and cellular processes such as dysregulated nutrient-sensing pathways, poor mitochondrial energy metabolism, inflammation, telomere shortening, and loss of tissue regeneration capacity that connect diabetes and aging are outlined in this study. The cardiovascular, renal, neurological, musculoskeletal, and endocrine systems are among the organ-specific signs of accelerated aging in T2DM that are highlighted. Knowing that T2DM is a condition of accelerated aging offers crucial information for managing and preventing the illness. In addition to good glycemic management and lifestyle change, targeting aging-related pathways may help prevent complications, maintain organ function, and enhance long-term health outcomes in people with T2DM.
Keywords
Insulin resistance, mitochondrial dysfunction, inflammation, type 2 diabetes mellitus, accelerated aging
Introduction
One of the biggest worldwide health issues of the twentyfirst century is type 2 diabetes mellitus (T2DM). According to the International Diabetes Federation (IDF), there are currently over 530 million people with diabetes globally, and by 2045, that number is predicted to rise to over 700 million [1]. Known as the “diabetes capital of the world,” India alone contributes significantly to this worldwide burden. Rapid urbanization, sedentary lifestyles, poor eating habits, and genetic predispositions are all blamed for this concerning trend [2]. Because T2DM is linked to cardiovascular disease, renal failure, and neurological problems, its socioeconomic effects extend beyond individuals to families and healthcare systems [3].
Traditionally, insulin resistance, β-cell dysfunction, and persistent hyperglycemia have been identified as the main characteristics of T2DM. Its pathogenesis has been closely associated with reduced physical activity, obesity, and excessive calorie consumption. Both macrovascular problems like atherosclerosis and coronary artery disease as well as microvascular problems like neuropathy, nephropathy, and retinopathy are brought on by prolonged exposure to hyperglycemia. Diabetic complications continue to appear earlier and more aggressively despite advancements in glucose control and medication, indicating that the disease process involves more than just metabolic dysregulation [4, 5].
A paradigm shift is supported by recent scientific data: T2DM is a condition of accelerated biological aging as well as a metabolic problem. Numerous studies have shown that people with T2DM exhibit early cellular and tissue aging, including telomere shortening, oxidative stress, mitochondrial dysfunction, and persistent lowgrade inflammation [6]. These processes coincide with the signs of aging, such as loss of proteostasis, chromosomal instability, and epigenetic changes. As a result, early-onset vascular stiffness, frailty, and cognitive decline—conditions usually associated with old age—are frequently seen in diabetic patients. be a result, T2DM is now referred to be a “metabolic disease of accelerated aging”[7].
The goal of this review is to investigate the complex biological and molecular connections between accelerated aging and T2DM. It aims to address the basic characteristics of aging and how they directly relate to the pathophysiology processes that underlie T2DM. The review continues to elucidate the molecular relationships between the two conditions, emphasizing oxidative stress, mitochondrial dysfunction, chronic inflammation, insulin resistance, telomere shortening, and epigenetic modifications. It also examines the organ-specific signs of accelerated aging seen in diabetic patients, such as early aging of the neurological, renal, and cardiovascular systems. Additionally, the review seeks to assess new therapeutic approaches that target aging-related pathways in diabetes management and to highlight recent developments in the identification of biomarkers of biological aging. This study offers a thorough and cohesive framework for understanding T2DM as both a metabolic problem and an aging disease, with important implications for personalized medication and public health initiatives. It does this by combining viewpoints from metabolic research and geroscience.
Conceptual framework for aging and metabolic dysfunction
Aging is a progressive and irreversible biological process characterized by the cumulative accumulation of molecular and cellular damage, leading to functional decline and increased vulnerability to disease. Contemporary geroscience frameworks describe aging as a regulated process driven by conserved biological mechanisms collectively termed the hallmarks of aging. Updated models identify twelve interconnected hallmarks, including genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, dysregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, altered intercellular communication, chronic inflammation, impaired autophagy, and gut microbiome dysbiosis (Table 1).
Table 1.
Types of stem cells in anemones.
| No | Hallmark of aging | Mechanistic description | Relevance to metabolic aging / T2DM | References |
|---|---|---|---|---|
| 1 | Genomic instability | Accumulation of DNA damage, mutations, and chromosomal aberrations with age. | Hyperglycemia-induced oxidative stress accelerates DNA damage in pancreatic β-cells and vascular endothelium. | [8] |
| 2 | Telomere attrition | Shortening of telomeres limits cellular replication capacity. | Shortened telomeres in diabetic patients correlate with insulin resistance and vascular aging. | [9] |
| 3 | Epigenetic alterations | DNA methylation and histone modification changes affecting gene expression. | Accelerated “epigenetic clock” observed in T2DM suggests faster biological aging. | [10] |
| 4 | Loss of proteostasis | Decline in chaperone activity and autophagy causing protein aggregation. | Hyperglycemia and lipid peroxidation promote misfolded protein accumulation in diabetic tissues. | [11] |
| 5 | Deregulated nutrient sensing | Disruption of the sirtuin, mTOR, AMPK, and insulin/IGF-1 signaling pathways. | Insulin resistance and mTOR overactivation are central to both aging and diabetes. | [12] |
| 6 | Mitochondrial dysfunction | Impaired mitochondrial biogenesis and increased ROS generation. | Diabetic tissues show defective ATP production and enhanced oxidative stress. | [13] |
| 7 | Cellular senescence | Irreversible growth arrest and SASP-mediated inflammation. | Senescent adipocytes and endothelial cells impair insulin signaling and tissue repair. | [12] |
| 8 | Stem cell exhaustion | Decline in regenerative potential of progenitor cells. | Reduced β-cell and muscle stem cell renewal in T2DM parallels premature aging. | [14] |
| 9 | Altered intercellular communication | Chronic low-grade inflammation and disrupted signaling. | Inflammatory cytokines (IL-6, TNF-α) drive “inflammaging” and insulin resistance. | [15] |
| 10 | Chronic inflammation | Persistent systemic inflammation from immune dysregulation. | Overlaps with diabetic inflammation, worsening vascular and metabolic dysfunction. | [16] |
| 11 | Disabled macroautophagy | Impaired removal of damaged organelles. | Reduced autophagy in diabetic muscle and liver contributes to metabolic stress. | [17] |
| 12 | Dysbiosis of gut microbiota | Imbalance of gut microorganisms affecting metabolism. | Alters glucose metabolism and insulin sensitivity in aging and diabetes. | [8] |
Importantly, many of these hallmarks are now measurable through established and emerging biological aging biomarkers, such as telomere length, DNA methylation clocks, mitochondrial DNA integrity, inflammatory mediators, proteomic and metabolomic signatures, and senescence-associated factors. In T2DM, these biomarkers consistently demonstrate premature activation and accelerated progression of aging pathways, indicating that diabetes represents a state of accelerated biological aging rather than normal chronological aging. Hyperglycemia, insulin resistance, oxidative stress, and chronic inflammation act as upstream drivers that amplify aging hallmarks, resulting in earlier onset of age-related phenotypes across multiple organs.
Thus, integrating aging biomarkers within this conceptual framework provides a comprehensive perspective for understanding metabolic dysfunction in T2DM and highlights accelerated aging biomarkers as both indicators of disease severity and potential targets for therapeutic intervention.
Metabolic aging and its acceleration in T2DM
Normal aging is accompanied by a gradual decline in metabolic flexibility—the capacity to efficiently switch between glucose and lipid utilization—resulting in impaired energy homeostasis and reduced cellular resilience. Age-related increases in oxidative stress, mitochondrial inefficiency, chronic inflammation, and diminished insulin responsiveness collectively predispose older individuals to insulin resistance and metabolic disease. These metabolic alterations closely resemble the pathophysiology of T2DM, supporting the concept that diabetes represents a state of accelerated metabolic aging, in which aging-related mechanisms are activated earlier and progress more rapidly.
Glucose homeostasis and mitochondrial energy dysfunction
During physiological aging, insulin sensitivity progressively declines in skeletal muscle, liver, and adipose tissue due to impaired insulin receptor signaling and reduced GLUT4 (glucose transporter type 4) translocation. Concurrently, pancreatic β-cell mass and secretory capacity decrease as a result of cumulative oxidative damage and mitochondrial stress [18]. Mitochondrial DNA mutations, impaired oxidative phosphorylation, defective mitophagy, and downregulation of proliferator–activated receptor gamma coactivator-1α (PGC-1α) further reduce ATP production and increase reactive oxygen species (ROS) generation [19]. In T2DM, chronic hyperglycemia and insulin resistance markedly intensify these processes. Hyperglycemia-induced oxidative stress accelerates mitochondrial damage, impairs lipid oxidation, promotes ectopic lipid accumulation, and hastens β-cell exhaustion. This convergence amplifies insulin resistance and reinforces accelerated metabolic aging [20].
Adipose tissue remodeling, protein metabolism, and sarcopenia
Aging is associated with redistribution of adipose tissue from subcutaneous to visceral depots, promoting systemic inflammation and insulin resistance. This shift is accompanied by adipokine imbalance, characterized by elevated leptin and reduced adiponectin levels, contributing to inflammaging and lipotoxicity in muscle and liver tissues [21]. Simultaneously, reduced muscle protein synthesis and anabolic resistance—driven by impaired mTORC1 signaling and decreased essential amino acid utilization— lead to sarcopenia, decreased energy expenditure, and frailty [22]. In T2DM, insulin resistance and mitochondrial dysfunction further suppress protein synthesis and muscle regeneration, accelerating muscle loss and functional decline beyond that expected with chronological aging.
Immune–metabolic inflammation and cellular senescence
Aging is characterized by persistent immune activation and increased production of pro-inflammatory cytokines such as TNF-α and IL-6, which interfere with insulin signaling and increase oxidative stress, forming a metabolic– aging axis [23]. In T2DM, chronic exposure to hyperglycemia and elevated free fatty acids sustains activation of inflammatory pathways such as NF-κB, intensifying inflammaging, endothelial dysfunction, and tissue fibrosis [24]. The accumulation of senescent cells further links aging and diabetes. Senescent cells in pancreatic β-cells, liver, and adipose tissue adopt a senescence-associated secretory phenotype (SASP), releasing pro-inflammatory cytokines and matrix-degrading enzymes. Hyperglycemia accelerates this senescent burden, leading to premature β-cell dysfunction and tissue deterioration [25].
Epigenetic alterations and nutrient-sensing dysregulation
Epigenetic alterations and telomere shortening are central markers of biological aging and are significantly accelerated in T2DM. Chronic metabolic stress induces oxidative DNA damage, telomere attrition, and aberrant DNA methylation patterns that advance epigenetic aging clocks and alter gene expression related to glucose metabolism and inflammation [26]. Parallel to these changes, dysregulation of nutrient-sensing pathways—including insulin/IGF 1 signaling, AMP-activated protein kinase (AMPK), and mechanistic target of rapamycin (mTOR)—is observed in both aging and T2DM. Chronic nutrient excess promotes sustained mTORC1 activation, suppresses autophagy, and accelerates cellular damage, while reduced AMPK activity impairs metabolic adaptation, reinforcing metabolic inflexibility and cellular senescence [27].
Stem cell exhaustion and integrated acceleration of aging
Stem cell exhaustion limits tissue regeneration during aging and is further exacerbated in T2DM. Metabolic stress and chronic inflammation impair pancreatic progenitor cells, skeletal muscle satellite cells, and bone marrow– derived stem cells. Hyperglycemia-induced oxidative damage reduces self-renewal capacity, accelerating β-cell failure, sarcopenia, and vascular complications [28]. Taken together, normal aging and T2DM share deeply overlapping metabolic mechanisms; however, in diabetes these processes are prematurely activated and pathologically intensified. T2DM therefore represents a model of accelerated metabolic aging, in which age-related molecular and cellular dysfunctions converge to drive early-onset organ decline and increased disease burden.
Mechanistic links between T2DM and acceler- ated aging
Oxidative stress and mitochondrial dysfunction
Chronic hyperglycemia in T2DM markedly increases the production of ROS within cellular mitochondria, thereby accelerating molecular and cellular processes typically associated with biological aging [29]. Persistently elevated glucose levels overload the mitochondrial electron transport chain, leading to electron leakage and excessive ROS generation [30]. This oxidative burden causes mitochondrial DNA (mtDNA) damage, lipid peroxidation, impaired oxidative phosphorylation, and reduced adenosine triphosphate (ATP) production, culminating in diminished cellular energy reserves and early tissue dysfunction [31].
Notably, these mitochondrial alterations mirror hallmark features of physiological aging, in which antioxidant defense systems—such as superoxide dismutase (SOD), catalase, and glutathione peroxidase—progressively decline, mitochondrial biogenesis is reduced (via downregulation of regulators including PGC-1α and NRF1), and mitochondrial fusion–fission dynamics become dysregulated [32]. In T2DM, however, these aging-associated mitochondrial defects emerge earlier and with greater severity, supporting the concept that diabetes represents a state of accelerated mitochondrial aging rather than merely metabolic dysfunction [33] (Figure 1).
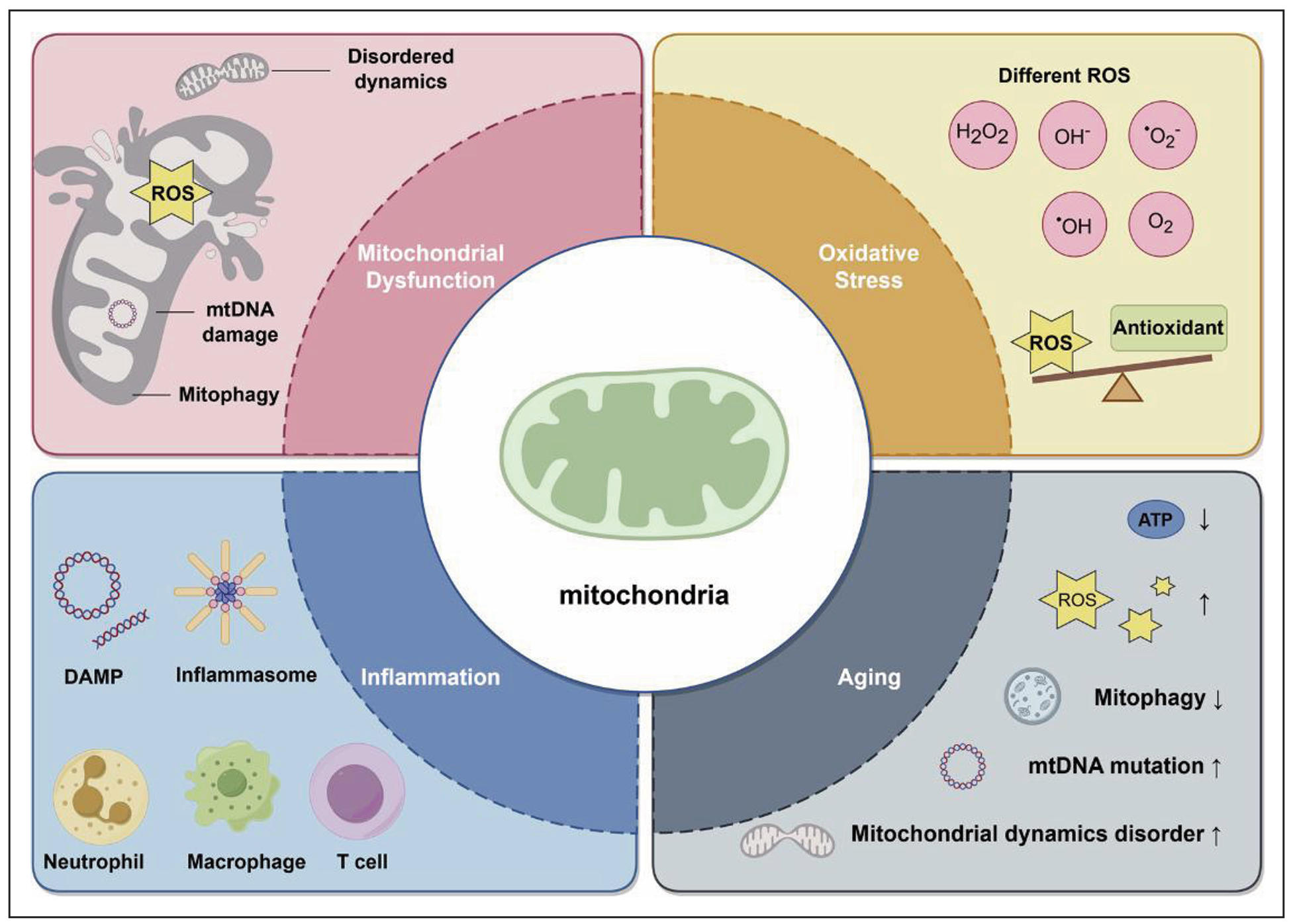
Figure 1. Mitochondrial dysfunction's crucial role in oxidative stress, inflammation, and aging.
Chronic inflammation
Aging is characterized by a persistent, low-grade inflammatory state termed inflammaging, which contributes to functional decline and age-related diseases. In T2DM, chronic metabolic stress—driven by hyperglycemia, elevated free fatty acids, and advanced glycation end products (AGEs)—sustains aberrant activation of proinflammatory pathways, including NF-κB signaling and the NLRP3 inflammasome. This results in persistently elevated levels of inflammatory mediators such as IL-6, TNF-α, and C-reactive protein (CRP), which impair insulin signaling, promote endothelial dysfunction, and accelerate tissue senescence [34].
While inflammaging also develops in non-diabetic aging, the inflammatory milieu in T2DM closely mimics—and markedly intensifies—this process, leading to premature immune dysregulation, impaired tissue repair, and cumulative organ damage. Thus, diabetes amplifies age-related inflammatory pathways, accelerating metabolic and vascular aging beyond what is observed in chronological aging alone (Table 2) [35].
Table 2.
Pro-inflammatory cytokines and their roles.
| Cytokine | Principal source | Role in aging | Role in T2DM | References |
|---|---|---|---|---|
| IL-6 | Adipose tissue/macrophages | Promotes inflammaging, muscle wasting | Insulin resistance, β-cell dysfunction | [36] |
| TNF-α | Activated macrophages | Inhibits cell repair, promotes senescence | Impairs insulin receptor signaling, endothelial damage | [37] |
| IL-1β | Inflammasome activation | Contributes to age-related immune dysregulation | β-cell apoptosis, adipose inflammation | [38] |
Advanced glycation end products
AGEs accumulate through the non-enzymatic glycation of proteins, lipids, and nucleic acids—a process that occurs slowly during normal aging but is substantially accelerated under hyperglycemic conditions. In T2DM, elevated glucose levels and reactive dicarbonyls such as methylglyoxal drive excessive AGE formation. Binding of AGEs to their receptor (RAGE) on endothelial cells, macrophages, and other tissues triggers ROS generation, NF-κB activation, pro-inflammatory cytokine release, and cellular senescence.
AGE-mediated cross-linking of collagen and extracellular matrix proteins reduces tissue elasticity, contributing to vascular stiffness and impaired organ function— phenotypes classically associated with advanced aging. Similarly, AGE accumulation during normal aging is linked to reduced cellular turnover, loss of proteostasis, and progressive tissue rigidity in organs such as the skin, kidneys, and vasculature. Therefore, the AGE–RAGE axis represents a critical mechanistic bridge between diabetes and aging, whereby metabolic dysregulation in T2DM accelerates molecular aging processes [39].
Insulin resistance and cellular senescence
Persistent insulin resistance is a defining feature of T2DM and a hallmark of metabolic aging. In both contexts, oxidative stress, chronic inflammation, and sustained nutrient excess impair insulin receptor signaling, particularly
along the PI3K–Akt–mTOR pathway, leading to reduced
glucose uptake and dysregulated lipid metabolism. These
metabolic insults simultaneously promote cellular senescence, an irreversible state of cell-cycle arrest accompanied by the SASP.
Senescent adipocytes, hepatocytes, and pancreatic β-cells secrete pro-inflammatory cytokines (including IL-6, IL8, and TNF-α), reinforcing local inflammation and further worsening insulin resistance. This establishes a selfperpetuating cycle in which diabetes accelerates agingrelated senescence, while senescence exacerbates diabetic pathology. Consistent with accelerated aging, diabetic tissues exhibit increased expression of senescence markers such as p16INK4a, p21Cip1, and SA-β-galactosidase activity—patterns also observed in aged organs. Importantly, experimental clearance of senescent cells via senolytic therapies improves insulin sensitivity in diabetic models, underscoring senescence as a key mediator of diabetesdriven accelerated aging [40] (Figure 2).
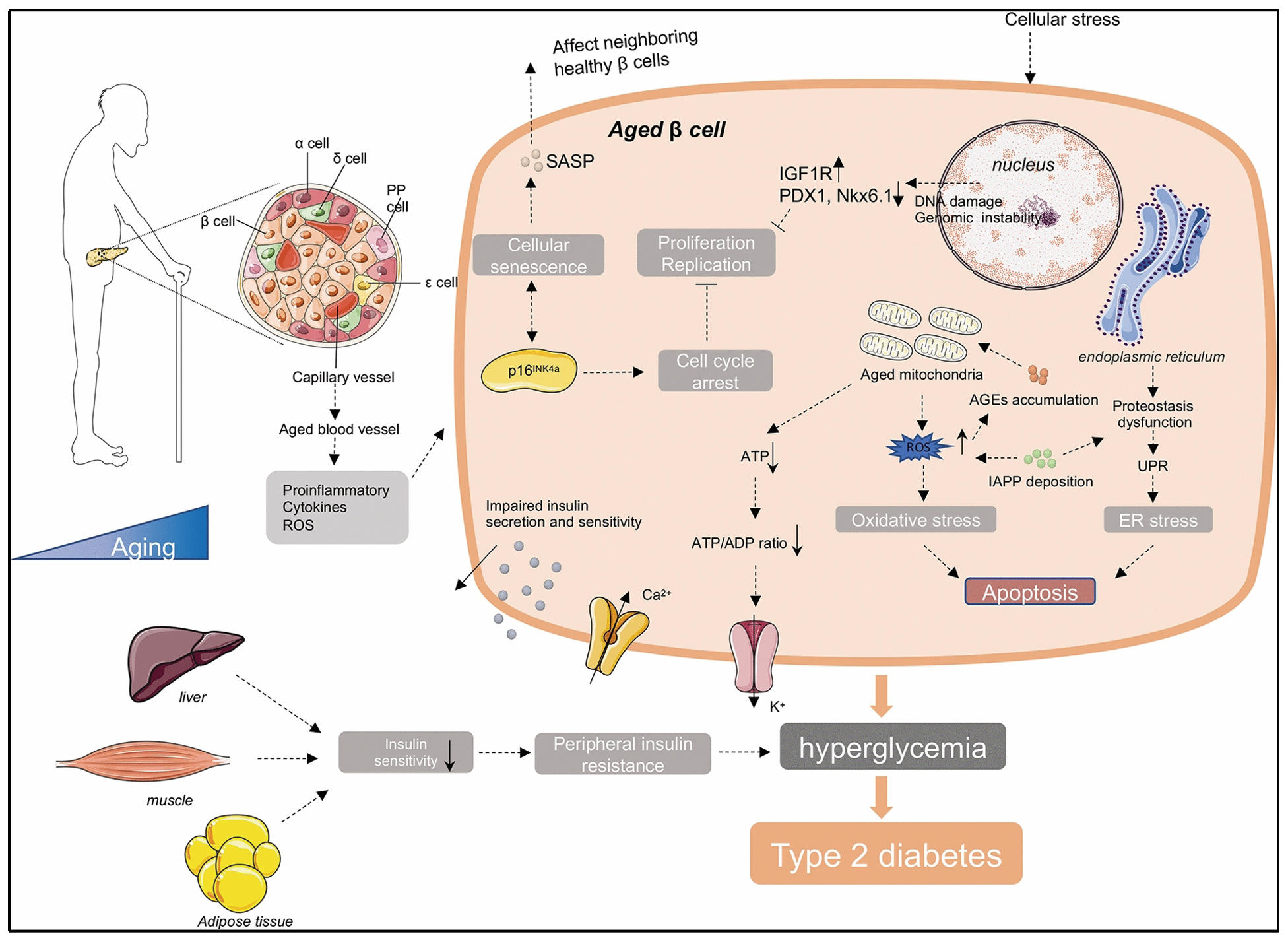
Figure 2. Aging-associated cellular stress and senescence in β-cell dysfunction leading to type 2 diabetes. Age-related mitochondrial dysfunction, oxidative and endoplasmic reticulum stress, impaired proteostasis, and SASP compromise insulin secretion and sensitivity, promoting hyperglycemia and systemic insulin resistance.
Genomic instability and telomere shortening
Telomeres, the repetitive DNA sequences that protect chromosome ends, progressively shorten with each cell division and are widely recognized as biomarkers of cellular aging. In T2DM, heightened oxidative stress and poor glycemic control are associated with accelerated telomere attrition, reflecting premature cellular aging. Hyperglycemia-induced ROS directly damage both genomic DNA and telomeric regions, while chronic inflammatory signaling further exacerbates telomere erosion.
Activation of DNA damage response (DDR) pathways, including p53, promotes senescence and apoptosis in vascular cells and pancreatic β-cells, contributing to early tissue dysfunction in diabetes [41]. These processes closely resemble those observed in natural aging, where accumulated DNA lesions, impaired repair mechanisms (e.g., PARP and ATM dysfunction), and telomere failure drive progressive cellular decline (Table 3). Thus, T2DM accelerates genomic aging by intensifying mechanisms normally associated with chronological aging.
Table 3.
Comparative overview of telomere dynamics in aging and T2DM.
| Parameter | Normal aging | Type 2 diabetes mellitus | References |
|---|---|---|---|
| Telomere length | Gradual shortening over decades | Rapid shortening due to chronic oxidative stress | [41] |
| DDR activation | Moderate, progressive | Persistent, exaggerated | [42] |
| Consequence | Slow cellular senescence | Premature senescence and apoptosis | [43] |
| Clinical impact | Age-related organ decline | Early vascular aging, neuropathy, β-cell loss | [44] |
Epigenetic changes
Epigenetic alterations—including DNA methylation, histone modifications, and non-coding RNA regulation—are fundamental mechanisms underlying physiological aging, contributing to progressive cellular dysfunction over time. Normal aging is characterized by a gradual acceleration of the epigenetic clock, marked by global DNA hypomethylation alongside site-specific hypermethylation of stressresponse and metabolic genes, leading to altered gene expression, mitochondrial decline, and increased inflammatory signaling.
In T2DM, these same epigenetic aging mechanisms are prematurely activated and intensified by chronic hyperglycemia. Aberrant DNA methylation of key metabolic and mitochondrial regulators—such as PPARGC1A, INS, and SIRT1—drives insulin resistance and mitochondrial dysfunction and gives rise to the phenomenon of “metabolic memory,” whereby aging-like cellular phenotypes persist even after glycemic normalization. Consistent with accelerated aging, individuals with T2DM exhibit epigenetic ages that are approximately 3–5 years older than age-matched non-diabetic controls, indicating advanced biological aging.
Additional epigenetic dysregulation further reinforces this acceleration. Altered histone acetylation due to reduced SIRT1 activity and increased HDAC signaling exacerbates oxidative stress and inflammation, while diabetes- and aging-associated non-coding RNAs—particularly miR-34a and miR-146a—promote cellular senescence, apoptosis, and chronic inflammatory responses. Collectively, these findings demonstrate that T2DM does not merely coexist with epigenetic aging but actively accelerates epigenetic aging trajectories, contributing to premature metabolic and organ dysfunction [45].
Organ-specific signs of T2DM-related accelerated aging
T2DM is increasingly recognized as a systemic disorder that accelerates aging-like changes across multiple organ systems. Chronic hyperglycemia, oxidative stress, mitochondrial dysfunction, and sustained inflammatory signaling converge to drive premature cellular senescence, fibrosis, and functional decline, recapitulating the molecular and structural features of physiological aging but occurring earlier and with greater severity. Thus, diabetes can be conceptualized as a model of accelerated biological aging rather than isolated metabolic disease.
In the cardiovascular system, T2DM induces premature vascular aging, characterized by endothelial dysfunction, arterial stiffness, and accelerated atherosclerosis. Chronic hyperglycemia promotes excessive formation of AGEs, which cross-link collagen and elastin within the vascular wall, reducing elasticity in a manner analogous to age-related arterial stiffening. Concurrently, reduced endothelial nitric oxide synthase (eNOS) activity limits nitric oxide (NO) bioavailability, impairing vasodilation and promoting vascular aging [46].
Oxidative stress and pro-inflammatory signaling (TNF-α, IL-6, NF-κB) further accelerate endothelial senescence and vascular smooth muscle cell proliferation, hastening plaque formation and arterial rigidity. These changes closely resemble those observed in aged vasculature but emerge decades earlier in individuals with diabetes, underscoring the concept of diabetes-driven cardiovascular aging.
Similarly, the kidneys of patients with T2DM display structural and functional alterations that parallel age-related nephrosclerosis. Chronic hyperglycemia induces glomerular basement membrane thickening, podocyte loss, and tubulointerstitial fibrosis—hallmark features of renal aging. Accelerated telomere shortening, mitochondrial dysfunction, and accumulation of senescent glomerular and tubular cells further impair renal repair capacity. Activation of senescence pathways, particularly the p16INK4a– p21Cip1 axis, enforces permanent cell-cycle arrest, limiting regeneration and promoting early decline in renal function [47] (Table 4). Collectively, these findings indicate that diabetic nephropathy represents a form of premature renal aging.
Table 4.
Comparative markers of renal aging and diabetic nephropathy
| Parameter | Aging kidney | Diabetic kidney | References |
|---|---|---|---|
| Glomerular morphology | Mild sclerosis | Nodular (Kimmelstiel–Wilson) sclerosis | [48] |
| Oxidative stress | Moderate | Markedly elevated | [49] |
| Mitochondrial function | Reduced ATP production | Severe mitochondrial loss | [50] |
| Senescence markers (p16, p21) | Increased | Strongly overexpressed | [51] |
| Repair capacity | Gradual decline | Early exhaustion of progenitor cells | [52] |
Neurodegeneration in T2DM likewise reflects accelerated neuronal aging. Insulin resistance within the brain, coupled with microvascular dysfunction, disrupts neuronal metabolism and synaptic plasticity. Glucose toxicity, mitochondrial failure, and chronic neuroinflammation (IL1β, TNF-α) promote neuronal loss and cognitive impairment. Clinical and imaging studies reveal increased tau hyperphosphorylation, amyloid-β accumulation, and faster cognitive decline in individuals with diabetes—features characteristic of Alzheimer’s disease, often described as “type 3 diabetes”. Chronic inflammatory exposure and oxidative mitochondrial DNA damage drive astrocyte and microglial senescence, further advancing neurodegenerative aging processes.
Skeletal muscle aging is also markedly accelerated in T2DM. Sarcopenia, a classical hallmark of aging, develops earlier and progresses more rapidly in diabetic individuals. Persistent inflammation, impaired insulin–Akt– mTOR signaling, and mitochondrial dysfunction reduce protein synthesis and muscle regeneration. Insulin resistance in skeletal muscle diminishes glucose uptake and ATP production, leading to muscle fatigue and atrophy. Diabetic neuropathy further exacerbates muscle loss by reducing motor unit activation. Combined with reduced physical activity and heightened oxidative stress, these mechanisms substantially increase frailty risk in diabetes, mirroring advanced biological aging [53] (Table 5).
Table 5.
Mechanistic comparison of age-related and diabetic sarcopenia.
| Mechanism | Normal aging | T2DM-associated | References |
|---|---|---|---|
| Insulin signaling | Gradual decline | Strong inhibition | [54] |
| Mitochondrial ROS | Moderate | Severe | [55] |
| Muscle stem cell activity | Reduced | Early depletion | [56] |
| Protein synthesis | Decreased | Impaired (via mTOR/AMPK imbalance) | [57] |
| Clinical effect | Sarcopenia | Sarcopenic obesity & frailty | [58] |
Within the endocrine pancreas, pancreatic β-cells exhibit hallmarks of accelerated cellular aging in T2DM. Prolonged exposure to glucotoxicity and lipotoxicity induces oxidative DNA damage, mitochondrial dysfunction, and activation of senescence pathways involving p53, p21, and p16. Senescent β-cells demonstrate impaired insulin secretion, increased release of SASP factors (including IL-6 and MCP-1), and diminished regenerative capacity. Experimental studies show that selective elimination of senescent β-cells via senolytic therapies restores insulin secretion and improves glucose homeostasis in diabetic models, highlighting β-cell aging as a central driver of metabolic dysfunction in T2DM [59].
Biological aging biomarkers in T2DM
Biological aging in T2DM is reflected by molecular biomarkers that indicate premature cellular and genomic decline. These aging signatures correlate strongly with vascular complications, cognitive impairment, frailty, and β-cell dysfunction, demonstrating that diabetes is associated with accelerated biological aging rather than normal chronological aging [60].
Telomere-based biomarkers
Telomeres shorten naturally with age, but in T2DM this process is markedly accelerated due to chronic hyperglycemia, oxidative stress, and persistent inflammation. Leukocyte telomere length (LTL) is significantly shorter in individuals with T2DM compared with age-matched nondiabetic controls, indicating an advanced biological age. Accelerated telomere attrition is closely linked to poor glycemic control (HbA1c) and activates DNA damage response pathways (p53/p21), promoting early cellular senescence in β-cells, endothelial cells, and renal cells. Reduced telomerase (hTERT) activity further limits telomere repair, reinforcing genomic instability and premature aging in diabetes [61] (Table 6).
Table 6.
Telomere Biomarkers in Aging vs. T2DM.
| Parameter | Normal Aging | T2DM Aging | References |
|---|---|---|---|
| Telomere length | Slow decline | Rapid shortening | [62] |
| Oxidative stress effect | Moderate | Severe, linked with hyperglycaemia | [63] |
| DDR activation | Low–moderate | High (↑p53, ↑γ-H2AX) | [64] |
| Functional outcome | Gradual senescence | Premature senescence, organ dysfunction | [65] |
Epigenetic biomarkers
Epigenetic dysregulation in T2DM reflects accelerated biological aging, driven by chronic hyperglycemia and metabolic stress. Aberrant DNA methylation of key aging- and metabolism-related genes (SIRT1, PPARGC1A, INS, GLUT4) and persistent “metabolic memory” promote inflammation, mitochondrial dysfunction, and premature cellular senescence [66]. DNA methylation clocks consistently show an epigenetic age acceleration of ~3–6 years in individuals with T2DM, strongly linking diabetes to accelerated aging [67, 68].
Mitochondrial Biomarkers
Mitochondrial biomarkers provide direct evidence of accelerated mitochondrial aging in T2DM. Reduced mtDNA copy number, increased oxidative mtDNA damage, impaired mitochondrial dynamics (↓MFN1/2, ↑DRP1), and electron transport chain dysfunction lead to reduced ATP production and early metabolic failure [69, 70]. These changes parallel age-related mitochondrial decline but occur earlier and more severely in diabetes, contributing to sarcopenia, fatigue, and β-cell dysfunction (Table 7).
Table 7.
Mitochondrial aging markers in T2DM
Biomarkers of oxidative stress and inflammation
Chronic low-grade inflammation (inflammaging) is a shared hallmark of physiological aging and T2DM, but in diabetes this process is accelerated and amplified, driving premature tissue dysfunction. Elevated circulating inflammatory markers—including IL-6, TNF-α, and CRP/ hs-CRP—are strongly associated with diabetic vascular aging, endothelial dysfunction, and insulin resistance, indicating advanced biological age. Increased levels of MCP-1, ICAM-1, and VCAM-1 further reflect premature endothelial activation and immune cell recruitment, key features of vascular aging.
Oxidative stress biomarkers provide complementary evidence of accelerated molecular aging in T2DM. Elevated 8-hydroxy-2′-deoxyguanosine (8-OHdG), malondialdehyde (MDA), and 8-iso-prostaglandin F2α (8-iso-PGF2α) indicate excessive oxidative DNA and lipid damage, while reduced antioxidant enzyme activity (GPx, catalase, SOD) reflects diminished stress resilience—hallmarks of advanced biological aging [76].
Proteomic and metabolomic biomarkers of accelerated aging
Metabolomic and proteomic profiling provides strong molecular evidence that T2DM is associated with premature biological aging.
Metabolomic biomarkers in T2DM mirror aging-related metabolic decline, including accumulation of branchedchain amino acids (BCAAs), excessive AGEs, and reduced NAD⁺ availability. Elevated BCAAs activate mTOR signaling, promote insulin resistance, and accelerate cellular senescence, while AGE accumulation drives vascular stiffness and chronic inflammation via RAGE– NF-κB signaling. A reduced NAD⁺/NADH ratio impairs sirtuin activity, mitochondrial biogenesis, and DNA repair, reinforcing metabolic and cellular aging pathways [77].
Proteomic biomarkers reveal a pro-senescent systemic environment in T2DM. Increased expression of SASP proteins (IL-6, IL-8, MMP-3, PAI-1) sustains inflammaging and accelerates vascular aging. Concurrent reductions in anti-aging proteins such as Klotho, SIRT1, and FGF21 reflect impaired stress resistance, mitochondrial dysfunction, and metabolic inflexibility—features characteristic of advanced biological aging. Increased glycated albumin further indicates cumulative metabolic damage and loss of proteostasis, closely paralleling age-related protein dysfunction [78].
Therapeutic implications include
Calorie restriction and intermittent fasting
Calorie restriction (CR) and intermittent fasting are among the most extensively studied non-pharmacological interventions targeting conserved aging pathways and metabolic health. At the molecular level, CR suppresses mTOR signaling, enhances autophagy, and reduces oxidative stress and chronic inflammation while activating key energy-sensing pathways such as AMPK and sirtuins.
These adaptations collectively lower nutrient-sensing signals, including insulin and insulin-like growth factor-1 (IGF-1), thereby promoting cellular maintenance and stress resistance. In the context of T2DM, CR improves insulin sensitivity, reduces ectopic lipid accumulation, attenuates adipose tissue inflammation, and mitigates metabolic dysfunctions associated with accelerated biological aging. Robust evidence from animal models demonstrates significant lifespan and healthspan extension with CR, whereas human clinical trials consistently report improvements in metabolic parameters and aging-related biomarkers. However, despite these promising findings, the long term impact of CR and intermittent fasting on human longevity remains to be fully established, underscoring the need for large-scale, long-duration intervention studies [79].
Organized training
Organized or structured physical training represents a cornerstone intervention for targeting both metabolic dysfunction and biological aging in T2DM. At the molecular level, regular exercise enhances insulin signaling efficiency, promotes skeletal muscle hypertrophy, and stimulates mitochondrial biogenesis and function through upregulation of peroxisome PGC-1α. These adaptations improve oxidative capacity, reduce ROS production, and preserve proteostasis, thereby counteracting sarcopenia and age-related mitochondrial decline. Clinically, organized training significantly reduces insulin resistance, improves glycemic control, and attenuates frailty, directly addressing two fundamental hallmarks of aging—loss of proteostasis and mitochondrial dysfunction. Owing to its robust and reproducible benefits, structured exercise training is strongly recommended in clinical guidelines for the management of T2DM, with additional potential to slow metabolic aging and extend healthspan [80].
Circadian synchronization (time-restricted feeding,sleep scheduling)
Circadian synchronization through time-restricted feeding and sleep scheduling improves metabolic health by aligning the body’s peripheral clocks—such as those in the liver and adipose tissue—with the central clock in the brain [81]. This alignment enhances glucose homeostasis by optimizing insulin sensitivity and glucose metabolism, while also reducing metabolic inflammation by regulat ing inflammatory pathways that are sensitive to circadian timing [82]. By restricting food intake to the active phase and maintaining regular sleep patterns, these behaviors act as strong time cues that coordinate metabolic processes across tissues, promoting more efficient energy utilization and glycemic control. This synchronization not only supports better management of blood glucose levels but also helps mitigate the physiological stresses associated with aging [83]. In contrast, when circadian rhythms are misaligned—due to irregular eating schedules, late-night feeding, or disrupted sleep—there is a breakdown in this coordination, leading to impaired glucose tolerance, increased inflammation, and accelerated metabolic dysfunction. Thus, aligning feeding and activity windows with the body’s natural circadian rhythm is a vital strategy to enhance metabolic health, prevent disease progression, and reduce age-related metabolic stress, as highlighted in studies summarized in Table 8 [84].
Table 8.
Pharmacological agents and their potential benefits in T2DM aging
| Intervention | Mechanism(s) relevant to aging | Potential benefits in T2DM aging | References |
|---|---|---|---|
| Metformin | Activates AMPK, inhibits mTOR signaling, reduces mitochondrial ROS, modulates epigenetic marks, promotes autophagy. | May reduce inflammation, vascular aging, counter risk signals; improves metabolic profile. | [85, 86] |
| SGLT2 inhibitors | Lowers glucotoxicity; improves cardiac/renal energetics; emerging data suggest reduced cellular senescence markers. | Reduces HF, CKD progression — major contributors to age-related morbidity; may attenuate senescence pathways. | [87, 88] |
| GLP-1 receptor agonists (GLP-1 RAs) | Weight loss, improved insulin sensitivity, anti-inflammatory effects; possible direct neuroprotective and multi-organ benefits. | By reducing obesity, inflammation and metabolic stress, may slow aging phenotypes; potential cognitive benefits reported. | [87, 89] |
| Senolytics (e.g., dasatinib + quercetin) | Selectively eliminate senescent cells (cellular senescence) — mitigate SASP; lower tissue inflammation | Direct targeting of fundamental aging mechanisms (kidney, vasculature) in carefully | [90, 91] |
| mTOR inhibitors / rapalogs (sirolimus / everolimus) | mTOR inhibition → promote cellular stress resistance. | Attenuate nutrient-signaling driven aging; theoretical metabolic benefit if | [92, 93] |
| NAD⁺ precursors (NR, NMN) | Restore NAD⁺ levels → support sirtuin activity, mitochondrial function, DNA repair. | Potential to improve mitochondrial dysfunction and metabolic resilience in T2DM. | [94] |
| Epigenetic therapies (HDAC/DNMT modulators, reprogramming approaches) | Reverse maladaptative epigenetic marks (age-related methylation), partial cellular reprogramming may reset aging signatures. | Could theoretically reverse aging signatures in metabolic tissues (pancreas, liver) — promising but experimental. | [95, 96] |
Future prospective
The focus of future research on T2DM must change from treating it as a metabolic disease to comprehending its intricate relationship to biological aging. According to geroscience-based models, metabolic degradation and aging are caused by processes such oxidative stress, cellular senescence, chronic inflammation, and mitochondrial malfunction. Thus, comprehensive strategies that incorporate lifestyle medicine, molecular aging biology, and endocrinology are crucial. Using indicators like mitochondrial indices, inflammatory signals, and epigenetic clocks, these models would enable the early identification of “accelerated aging phenotypes” in diabetic patients. Interventions that simultaneously enhance glycemic management and reduce the underlying aging process will be found thanks to this convergence of fields will improve the basis for gerotherapeutics in diabetes to be approved by regulators.
Conclusions
It is becoming more well acknowledged that T2DM is both a metabolic disease and a syndrome marked by accelerated biological aging. Evidence from molecular biology, clinical research, and biomarker studies has come together to show that pathways like oxidative stress, cellular senescence, mitochondrial failure, and chronic inflammation are common mechanisms that drive the course of diabetes and aging. A more thorough framework for explaining the early onset of problems, decreased physiological resilience, and increased vulnerability seen in diabetics is provided by an understanding of this reciprocal interaction. The management of diabetes in the future will be significantly impacted by this aging-centric viewpoint. Therapeutic approaches must address the deeper biological mechanisms controlling aging itself rather than concentrating only on glycemic management. Metformin, GLP-1 agonists, SGLT2 inhibitors, senolytics, and NAD+ -enhancing agents are among the pharmacological strategies that have the potential to slow both metabolic decline and the aging trajectory when paired with lifestyle interventions like structured exercise, calorie restriction, and circadian alignment. Clinicians may be able to customize treatments based on biological age rather than chronological age by incorporating aging biomarkers into routine evaluation. All things considered, redefining T2DM as a metabolic disease of accelerated aging creates new opportunities for clinical translation and innovation in public health. Healthcare systems can transition from reactive disease control to proactive healthspan extension by connecting geroscience and diabetology. In addition to lowering long-term problems, this strategy may enhance the general quality of life and functional ability of people with T2DM. In the end, this integrated approach might turn diabetes care into a field that emphasizes longevity, resilience, and prevention.
Declarations
Authorship Contributions
Simran C. Yadav: Conceptualization, investigation, writing original draft. Shraddha V. Mahale: conceptualization, investigation, writing original draft. Sneha M. Nishad: resources, data curation, visualization, formal analysis. Bhumika D. Bankar: resources, data curation, visualization, formal analysis. Pooja B. Rasal: resources, data curation, visualization, formal analysis.
Financial support and sponsorship
None.
Conflict of interest
The authors confirm that there are no known conflicts of interest.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
References
1. Pradeepa R, & Mohan V. Epidemiology of type 2 diabetes in India. Indian J Ophthalmol, 2021, 69(11): 2932-2938. [Crossref]
2. Khan M, Hashim M, King J, Govender R, Mustafa H, & Al Kaabi J. Epidemiology of type 2 diabetes - global burden of disease and forecasted trends. J Epidemiol Glob Health, 2020, 10(1): 107-111. [Crossref]
3. Vidit K, & Syed S. Epidemiology and prevalence of type 2 diabetes mellitus in children with obesity. European Journal of Medical and Health Sciences, 2021, 3(1): 3943. [Crossref]
4. Laslett L, Alagona P, Jr., Clark B, 3rd, Drozda J, Jr., Saldivar F, Wilson S, et al. The worldwide environment of cardiovascular disease: prevalence, diagnosis, therapy, and policy issues: a report from the American College of Cardiology. J Am Coll Cardiol, 2012, 60(25 Suppl): S1-49. [Crossref]
5. Hameed I, Masoodi S, Mir S, Nabi M, Ghazanfar K, & Ganai B. Type 2 diabetes mellitus: from a metabolic disorder to an inflammatory condition. World J Diabetes, 2015, 6(4): 598-612. [Crossref]
6. Bahour N, Cortez B, Pan H, Shah H, Doria A, & AguayoMazzucato C. Diabetes mellitus correlates with increased biological age as indicated by clinical biomarkers. Geroscience, 2022, 44(1): 415-427. [Crossref]
7. Wölfel E, & Kassem M. New aspects of the pathophysiology of diabetic bone fragility - type 2 diabetes mellitus as a disease of accelerated aging. Curr Osteoporos Rep, 2025, 23(1): 45-57. [Crossref]
8. López-Otín C, Blasco M, Partridge L, Serrano M, & Kroemer G. The hallmarks of aging. Cell, 2013, 153(6): 1194-1217. [Crossref]
9. Rasal P, Kasar G, Pagar D, Upaganlawar A, & Mahajan SK. Resilience in the depths: anemones as models for aging and regeneration research. Aging Pathobiol Ther, 2015, 7(3): 144-155. [Crossref]
10. Skowronska-Krawczyk D. Hallmarks of aging: causes and consequences. Aging Biol, 2023, 1(1): 20230011. [Crossref]
11. Colchero F, Aburto J, Archie E, Boesch C, Breuer T, Campos F, et al. The long lives of primates and the ‘invariant rate of ageing’ hypothesis. Nat Commun, 2021, 12(1): 3666. [Crossref]
12. Baechle J, Chen N, Makhijani P, Winer S, Furman D, & Winer D. Chronic inflammation and the hallmarks of aging. Mol Metab, 2023, 74: 101755. [Crossref]
13. López-Otín C, Blasco M, Partridge L, Serrano M, & Kroemer G. Hallmarks of aging: an expanding universe. Cell, 2023, 186(2): 243-278. [Crossref]
14. Kaushik S, Tasset I, Arias E, Pampliega O, Wong E, Martinez-Vicente M, et al. Autophagy and the hallmarks of aging. Ageing res rev, 2021, 72: 101468. [Crossref]
15. Liu G, & Sabatini D. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol, 2020, 21(4): 183-203. [Crossref]
16. Ackermann M, Chao L, Bergstrom C, & Doebeli M. On the evolutionary origin of aging. Aging cell, 2007, 6(2): 235-244. [Crossref]
17. Dominguez L, Veronese N, & Barbagallo M. Magnesium and the hallmarks of aging. Nutrients, 2024, 16(4): 496505. [Crossref]
18. Zhang K, Ma Y, Luo Y, Song Y, Xiong G, Ma Y, et al. Metabolic diseases and healthy aging: identifying environmental and behavioral risk factors and promoting public health. Front Public Health, 2023, 11: 1253506. [Crossref]
19. Kolb H, Kempf K, & Martin S. Insulin and aging - a disappointing relationship. Front Endocrinol, 2023, 14: 1261298. [Crossref]
20. Oyewande A, Iqbal B, Abdalla L, Karim F, & Khan S. An Overview of the pathophysiology of metabolic changes and their sequence of occurrence in obese diabetic females: a narrative review. Cureus, 2020, 12(10): e10947. [Crossref]
21. Palmer A, & Jensen M. Metabolic changes in aging humans: current evidence and therapeutic strategies. J Clin Invest, 2022, 132(16): e158451. [Crossref]
22. Zhou Q, Yu L, Cook J, Qiang L, & Sun L. Deciphering the decline of metabolic elasticity in aging and obesity. Cell Metab, 2023, 35(9): 1661-1671.e1666. [Crossref]
23. Moldakozhayev A, & Gladyshev V. Metabolism, homeostasis, and aging. Trends Endocrinol Metab, 2023, 34(3): 158-169. [Crossref]
24. Wikström Shemer D, Mostafaei S, Tang B, Pedersen N, Karlsson I, Fall T, et al. Associations between epigenetic aging and diabetes mellitus in a Swedish longitudinal study. Geroscience, 2024, 46(5): 5003-5014. [Crossref]
25. Zong Y, Li H, Liao P, Chen L, Pan Y, Zheng Y, et al. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct Target Ther, 2024, 9(1): 124136. [Crossref]
26. Chakrabarti S, & Chattopadhyay D. The Link between immune aging and type 2 diabetes: a review of mechanisms and implications. Exploratory Research and Hypothesis in Medicine, 2025, 10(3): 185-204. [Crossref]
27. Odimegwu C, Uwaezuoke S, Chikani U, Mbanefo N, Adiele K, Nwolisa C, et al. Targeting the epigenetic marks in type 2 diabetes mellitus: will epigenetic therapy be a valuable adjunct to pharmacotherapy? Diabetes Metab Syndr Obes, 2024, 17: 3557-3576. [Crossref]
28. Chang X, & Lin W. Epigenetic age acceleration mediates the association between smoking and diabetes-related outcomes. Clin Epigenetics, 2023, 15(1): 94109. [Crossref]
29. Yang T, Qi F, Guo F, Shao M, Song Y, Ren G, et al. An update on chronic complications of diabetes mellitus: from molecular mechanisms to therapeutic strategies with a focus on metabolic memory. Mol Med, 2024, 30(1): 71-88. [Crossref]
30. Caturano A, D’Angelo M, Mormone A, Russo V, Mollica M, Salvatore T, et al. Oxidative stress in type 2 diabetes: impacts from pathogenesis to lifestyle modifications. Curr Issues Mol Biol, 2023, 45(8): 6651-6666. [Crossref]
31. Kowluru R, Kowluru A, Mishra M, & Kumar B. Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog Retin Eye Res, 2015, 48: 40-61. [Crossref]
32. Cojocaru K, Luchian I, Goriuc A, Antoci L, Ciobanu C, Popescu R, et al. Mitochondrial dysfunction, oxidative stress, and therapeutic strategies in diabetes, obesity, and cardiovascular disease. Antioxidants, 2023, 12(3): 658-669. [Crossref]
33. Shi T, Zhou Z, Jiang W, Huang T, Si J, & Li L. Hyperglycemia-induced oxidative stress exacerbates mitochondrial apoptosis damage to cochlear stria vascularis pericytes via the ROS-mediated Bcl-2/CytC/AIF pathway. Redox Rep, 2024, 29(1): 2382943. [Crossref]
34. Aranda J, Ramírez C, & Mittelbrunn M. Inflammageing, a targetable pathway for preventing cardiovascular diseases. Cardiovasc Res, 2025, 121(10): 1537-1550. [Crossref]
35. Karamitsos K, Oikonomou E, Theofilis P, Ikonomidis I, Kassi E, Lambadiari V, et al. The role of NLRP3 inflammasome in type 2 diabetes mellitus and its macrovascular complications. J Clin Med, 2025, 14(13): 4606-4618. [Crossref]
36. Cao C, Yuan J, Gilbert E, Cline M, Lam F, Li K, et al. Increased circulating interleukin concentrations in type 2 diabetes: a systematic review and meta-analysis. Obes Rev, 2025, 26(12): e13971. [Crossref]
37. Gora I, Ciechanowska A, & Ladyzynski P. NLRP3 inflammasome at the interface of inflammation, endothelial dysfunction, and type 2 diabetes. Cells, 2021, 10(2): 314-328. [Crossref]
38. Iwasaki K, Abarca C, & Aguayo-Mazzucato C. Regulation of cellular senescence in type 2 diabetes mellitus: from mechanisms to clinical applications. Diabetes Metab J, 2023, 47(4): 441-453. [Crossref]
39. Ojeda-Rodriguez A, Rangel-Zuñiga O, Arenas-de Larriva A, Gutierrez-Mariscal F, Torres-Peña J, Romero-Cabrera J, et al. Telomere length as biomarker of nutritional therapy for prevention of type 2 diabetes mellitus development in patients with coronary heart disease: CORDIOPREV randomised controlled trial. Cardiovasc Diabetol, 2024, 23(1): 98-107. [Crossref]
40. Gavia-García G, Rosado-Pérez J, Arista-Ugalde T, Aguiñiga-Sánchez I, Santiago-Osorio E, & Mendoza-Núñez V. Telomere length and oxidative stress and its relation with metabolic syndrome components in the aging. Biology (Basel), 2021, 10(4): 253-264. [Crossref]
41. He X, Cao L, Fu X, Wu Y, Wen H, Gao Y, et al. The association between telomere length and diabetes mellitus: accumulated evidence from observational studies. J Clin Endocrinol Metab, 2024, 110(1): e177-e185. [Crossref]
42. Yang C, Huang M, Davis W, Jenkins A, & Davis T. Determinants of temporal change in telomere length and its associations with chronic complications and mortality in type 2 diabetes: the Fremantle diabetes study phase II. Cardiovasc Diabetol, 2025, 24(1): 267-278. [Crossref]
43. Kiełbowski K, Bakinowska E, & Pawlik A. Epigenetics plays a role in the pathogenesis and treatment of diabetes. Genes, 2025, 16(7): 769-779. [Crossref]
44. Lunder M, Janić M, & Šabovič M. Treating arterial ageing in patients with diabetes: from mechanisms to effective drugs. International Journal of Molecular Sciences, 2021, 22(6): 2796.
45. Flemming N, Pernoud L, Forbes J, & Gallo L. Mitochondrial dysfunction in individuals with diabetic kidney disease: a systematic review. Cells, 2022, 11(16): 2481-2496. [Crossref]
46. Phillips P, de Sousa Loreto Aresta Branco M, Cliff C, Ward J, Squires P, & Hills C. Targeting senescence to prevent diabetic kidney disease: exploring molecular mechanisms and potential therapeutic targets for disease management. Diabet Med, 2025, 42(2): e15408. [Crossref]
47. Jin Q, Ma F, Liu T, Yang L, Mao H, Wang Y, et al. Sirtuins in kidney diseases: potential mechanism and therapeutic targets. Cell Commun Signal, 2024, 22(1): 114. [Crossref]
48. Chi M, Tian Z, Ma K, Li Y, Wang L, Nasser M, et al. The diseased kidney: aging and senescent immunology. Immun Ageing, 2022, 19(1): 58-69. [Crossref]
49. Huang J, Pang D, Fan C, Yang G, Chen J, & Chen S. Integrative transcriptomic and machine learning analyses identify HDAC9 as a key regulator of mitochondrial dysfunction and senescence-associated inflammation in diabetic nephropathy. Frontiers in Immunology, 2025, Volume 16. [Crossref]
50. Zhang Y, Zhao Y, Liu Y, Fang Y, Sun L, Wei S, et al. High glucose induces renal tubular epithelial cell senescence by inhibiting autophagic flux. Hum Cell, 2025, 38(2): 4354. [Crossref]
51. Xie Z, Li Y, Li X, & Zhang J. A review of mitochondrial dysfunction in diabetic sarcopenia: mechanisms, diagnosis, and treatment approaches. J Int Med Res, 2025, 53(7): 3000605251355996. [Crossref]
52. Purnamasari D, Tetrasiwi E, Kartiko G, Astrella C, Husam K, & Laksmi P. Sarcopenia and chronic complications of type 2 diabetes mellitus. Rev Diabet Stud, 2022, 18(3): 157-165. [Crossref]
53. Shou J, Chen P, & Xiao W. Mechanism of increased risk of insulin resistance in aging skeletal muscle. Diabetol Metab Syndr, 2020, 12: 14-25. [Crossref]
54. Ortez Toro J. Diabetes and sarcopenia: unraveling the metabolic crossroads of muscle loss and glycemic dysregulation. Endocrines, 2025, 6(3): 47-58.
55. Cruz-Jentoft A, Baeyens J, Bauer J, Boirie Y, Cederholm T, Landi F, et al. Sarcopenia: European consensus on definition and diagnosis: report of the European working group on sarcopenia in older people. Age Ageing, 2010, 39(4): 412-423. [Crossref]
56. Sbrignadello S, Göbl C, & Tura A. Bioelectrical impedance analysis for the assessment of body composition in sarcopenia and type 2 diabetes. Nutrients, 2022, 14(9): 1864-1876. [Crossref]
57. Sodini S, & Suarez-Ortegón M. Senolytic interventions for type 2 diabetes: current evidence and future directions. Diabetology, 2025, 6(6): 48-60.
58. Ma D, Zhu W, Hu S, Yu X, & Yang Y. Association between oxidative stress and telomere length in type 1 and type 2 diabetic patients. J Endocrinol Invest, 2013, 36(11): 1032-1037. [Crossref]
59. Sawicki K, Matysiak-Kucharek M, Gorczyca-Siudak D, Kruszewski M, Kurzepa J, Kapka-Skrzypczak L, et al. Leukocyte telomere length as a marker of chronic complications in type 2 diabetes patients: a risk assessment study. Int J Mol Sci, 2024, 26(1): 290-301. [Crossref]
60. Dudinskaya E, Tkacheva O, Shestakova M, Brailova N, Strazhesko I, Akasheva D, et al. Short telomere length is associated with arterial aging in patients with type 2 diabetes mellitus. Endocr Connect, 2015, 4(3): 136-143. [Crossref]
61. Narasimhan A, Flores R, Robbins P, & Niedernhofer L. Role of cellular senescence in type II diabetes. Endocrinology, 2021, 162(10): bqab136. [Crossref]
62. Uziel O, Singer J, Danicek V, Sahar G, Berkov E, Luchansky M, et al. Telomere dynamics in arteries and mononuclear cells of diabetic patients: effect of diabetes and of glycemic control. Exp Gerontol, 2007, 42(10): 971-978. [Crossref]
63. Baliou S, Apetroaei M, Hatzidaki E, Kuzmin S, Tzatzarakis M, Arsene A, et al. The interplay between obesity and type 2 diabetes: common pathophysiological mechanisms contributing to telomere shortening. Life (Basel), 2025, 15(6): 873-884. [Crossref]
64. Ling C, & Rönn T. Epigenetics in human obesity and type 2 diabetes. Cell Metab, 2019, 29(5): 1028-1044. [Crossref]
65. Wang A, Wang S, Wang B, Xiao M, Guo Y, Tang Y, et al. Epigenetic regulation associated with sirtuin 1 in complications of diabetes mellitus. Front Endocrinol, 2020, 11: 598012. [Crossref]
66. Ahmed I, Chakraborty R, Faizy A, & Moin S. Exploring the key role of DNA methylation as an epigenetic modulator in oxidative stress related islet cell injury in patients with type 2 diabetes mellitus: a review. J Diabetes Metab Disord, 2024, 23(2): 1699-1718. [Crossref]
67. Sathishkumar C, Prabu P, Balakumar M, Lenin R, Prabhu D, Anjana R, et al. Augmentation of histone deacetylase 3 (HDAC3) epigenetic signature at the interface of proinflammation and insulin resistance in patients with type 2 diabetes. Clin Epigenetics, 2016, 8: 125-138. [Crossref]
68. Hu L, Ding M, Tang D, Gao E, Li C, Wang K, et al. Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics, 2019, 9(13): 3687-3706. [Crossref]
69. Wang X, Cui N, Liu X, & Liu X. Mitochondrial 8-hydroxy2′-deoxyguanosine and coronary artery disease in patients with type 2 diabetes mellitus. Cardiovascular Diabetology, 2020, 19(1): 22-34. [Crossref]
70. Wang S, Zhao H, Lin S, Lv Y, Lin Y, Liu Y, et al. New therapeutic directions in type II diabetes and its complications: mitochondrial dynamics. Front Endocrinol, 2023, 14: 1230168. [Crossref]
71. Van Huynh T, Rethi L, Rethi L, Chen C, Chen Y, & Kao Y. The complex interplay between imbalanced mitochondrial dynamics and metabolic disorders in type 2 diabetes. Cells, 2023, 12(9): 1223-1238. [Crossref]
72. Vezza T, Díaz-Pozo P, Canet F, de Marañón AM, AbadJiménez Z, García-Gargallo C, et al. The role of mitochondrial dynamic dysfunction in age-associated type 2 diabetes. World J Mens Health, 2022, 40(3): 399-411. [Crossref]
73. Wang W, Luo J, Willems van Dijk K, Hägg S, Grassmann F, LM T, et al. Assessment of the bi-directional relationship between blood mitochondrial DNA copy number and type 2 diabetes mellitus: a multivariable-adjusted regression and Mendelian randomisation study. Diabetologia, 2022, 65(10): 1676-1686. [Crossref]
74. Gilkerson R. Commentary: mitochondrial DNA damage and loss in diabetes. Diabetes Metab Res Rev, 2016, 32(7): 672-674. [Crossref]
75. López-Contreras A, Martínez-Ruiz M, Olvera-Montaño C, Robles-Rivera R, Arévalo-Simental D, CastellanosGonzález J, et al. Importance of the use of oxidative stress biomarkers and inflammatory profile in aqueous and vitreous humor in diabetic retinopathy. Antioxidants, 2020, 9(9): 891-906. [Crossref]
76. Fan L, Cacicedo J, & Ido Y. Impaired nicotinamide adenine dinucleotide (NAD+) metabolism in diabetes and diabetic tissues: implications for nicotinamide-related compound treatment. J Diabetes Investig, 2020, 11(6): 1403-1419. [Crossref]
77. Gu H, Jiang W, You N, Huang X, Li Y, Peng X, et al. Soluble klotho improves hepatic glucose and lipid homeostasis in type 2 diabetes. Mol Ther Methods Clin Dev, 2020, 18: 811-823. [Crossref]
78. Kulkarni A, Gubbi S, & Barzilai N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab, 2020, 32(1): 15-30. [Crossref]
79. Mohammed I, Hollenberg M, Ding H, & Triggle C. A critical review of the evidence that metformin is a putative anti-aging drug that enhances healthspan and extends lifespan. Front Endocrinol, 2021, 12: 718942. [Crossref]
80. Hu D, Xie F, Xiao Y, Lu C, Zhong J, Huang D, et al. Metformin: a potential candidate for targeting aging mechanisms. Aging Dis, 2021, 12(2): 480-493. [Crossref]
81. Chen S, Gan D, Lin S, Zhong Y, Chen M, Zou X, et al. Metformin in aging and aging-related diseases: clinical applications and relevant mechanisms. Theranostics, 2022, 12(6): 2722-2740. [Crossref]
82. La Grotta R, Frigé C, Matacchione G, Olivieri F, de Candia P, Ceriello A, et al. Repurposing SGLT-2 inhibitors to target aging: available evidence and molecular mechanisms. Int J Mol Sci, 2022, 23(20): 12325. [Crossref]
83. Shalaby Y, Nakhal M, Afandi B, Al-Zohily B, Majed L, Kumar K, et al. Impact of sodium-glucose cotransporter-2 inhibitors on aging biomarkers and plasma ceramide levels in type 2 diabetes: beyond glycemic control. Ann Med, 2025, 57(1): 2496795. [Crossref]
84. Kreiner F, von Scholten B, Kurtzhals P, & Gough S. Glucagon-like peptide-1 receptor agonists to expand the healthy lifespan: current and future potentials. Aging cell, 2023, 22(5): e13818. [Crossref]
85. Islam M, Tuday E, Allen S, Kim J, Trott D, Holland W, et al. Senolytic drugs, dasatinib and quercetin, attenuate adipose tissue inflammation, and ameliorate metabolic function in old age. Aging cell, 2023, 22(2): e13767. [Crossref]
86. Schweiger A, Diniz B, Nicol G, Schweiger J, Dasklakis-Perez A, & Lenze E. Protocol for a pilot clinical trial of the senolytic drug combination dasatinib plus quercetin to mitigate age-related health and cognitive decline in mental disorders. F1000Res, 2024, 13: 1072-1086. [Crossref]
87. Yanai H, Adachi H, Hakoshima M, & Katsuyama H. Adenosine monophosphate-activated protein kinase activation and mammalian target of rapamycin complex 1 inhibition: a mechanistic rationale for anti-aging therapy in type 2 diabetes. J Clin Med Res, 2025, 17(9): 469-489. [Crossref]
88. Barzilai N, Crandall J, Kritchevsky S, & Espeland M. Metformin as a tool to target aging. Cell Metab, 2016, 23(6): 1060-1065. [Crossref]
89. Prattichizzo F, De Nigris V, Mancuso E, Spiga R, Giuliani A, Matacchione G, et al. Short-term sustained hyperglycaemia fosters an archetypal senescence-associated secretory phenotype in endothelial cells and macrophages. Redox Biol, 2018, 15: 170-181. [Crossref]
90. Delrue C, Speeckaert R, & Speeckaert M. Rewinding the clock: emerging pharmacological strategies for human anti-aging therapy. Int J Mol Sci, 2025, 26(19): 9372. [Crossref]
91. Yesilyurt-Dirican Z, Qi C, Wang Y, Simm A, Deelen L, Hafiz Abbas Gasim A, et al. SGLT2 inhibitors as a novel senotherapeutic approach. NPJ Aging, 2025, 11(1): 35. [Crossref]
92. Cheng F, Liu Y, Du J, & Lin J. Metformin’s mechanisms in attenuating hallmarks of aging and age-related disease. Aging Dis, 2022, 13(4): 970-986. [Crossref]
93. Murillo-Cancho A, Lozano-Paniagua D, & Nievas-Soriano B. Dietary and pharmacological modulation of agingrelated metabolic pathways: molecular insights, clinical evidence, and a translational model. Int J Mol Sci, 2025, 26(19): 9643-9655. [Crossref]
94. Zhou Z, Duan H, Xue S, & Li Z. Progress in anti-ageing drug research for age-related diseases: a review. Ageing res rev, 2026, 114: 102982.[Crossref]
95. Witham M, Granic A, Pearson E, Robinson S, & Sayer A. Repurposing drugs for diabetes mellitus as potential pharmacological treatments for sarcopenia – a narrative review. Drugs Aging, 2023, 40(8): 703-719. [Crossref]
96. Katsuumi G, Shimizu I, Suda M, Yoshida Y, Furihata T, Joki Y, et al. SGLT2 inhibition eliminates senescent cells and alleviates pathological aging. Nat Aging, 2024, 4(7): 926–938. [Crossref]