Open Access | Review
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
A mechanistic narrative review of phytochemical enhancement of mitophagy in Parkinson’s disease: from PINK1/Parkin pathways to dopaminergic neuroprotection
* Corresponding author: Abraham Olufemi Asuku
Mailing address: Bioresources Development Centre, National Biotechnology Research and Development Agency, Ogbomoso, Nigeria.
Email: asufem2017@gmail.com
Received: 01 February 2026 / Revised: 28 February 2026 / Accepted: 13 March 2026 / Published: 31 March 2026
DOI: 10.31491/APT.2026.03.204
Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative disorder caused by the loss of dopaminergic neurons, leading to manifestations of motor and non-motor dysfunctions. Mitochondrial dysfunction and impaired mitophagy contribute to the development and progression of PD, underlying dopaminergic neurodegeneration. The PINK1/Parkin signalling pathway is a cellular quality control system contributing to dysfunctional mitochondria clearance. Its dysregulation is a canonical mechanism in both sporadic and familial PD pathogenesis. Phytochemicals have been reported to restore PINK1/Parkin signalling pathway and improve neuronal health by maintaining mitochondrial homeostasis and mitophagy cascade. This review underscores the mechanisms of mitophagy modulation by phytochemicals through PINK1/Parkin signalling pathway and critically synthesize current and associated molecular, biochemical, and functional networks, reinforcing their significance to dopaminergic neuroprotection in PD experimental models. Studies published between 2010 and 2025 were comprehensively searched across Google scholar and PubMed. Studies examining the effects of phytochemicals on mitochondrial function, mitophagy, and PINK1/Parkin signalling pathways in experimental PD models were the central focus of this review. Phytonutrients including baicalein, berberine, and resveratrol promote mitochondrial homeostasis and dopaminergic neuroprotection by enhancing mitophagy through PINK1 stabilization, Parkin translocation and modulation of Nrf2 and SIRT1 pathways. Despite the promising neuroprotective effects of these phytonutrients, limitations including bioavailability, dosage, and experimental designs are major drawbacks necessitating standardized studies and clinical validation to make these findings translationally therapeutic for human with PD. Modulatory effects of phytoch
Keywords
Parkinson’s disease, PINK1/Parkin, mitophagy, mitochondrial homeostasis, phytochemicals, dopaminergic neuroprotection
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disease associated with the death or loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc). Nigrostriatal loss of dopamine in PD leads to manifestation of motor dysfunction including bradykinesia, rigidity, tremor, and postural instability [1]. Asides motor dysfunctions in PD, there are non-motor symptoms of PD including pain, hyposmia, sleep disorders, depression and constipation which sometime precede the clinical manifestations of PD for several years [2]. Both motor and non-motor symptoms of PD manifest across the spectrum of PD patients, including familial and sporadic PD. Most treatment options in PD are symptomatic treatments with restoration of dopaminergic tone in the nigrostriatal axis. However, the current treatments do not address the underlying mechanisms driving dopaminergic neuronal loss [3].
Many factors including neuroinflammation, protein misfolding, oxidative stress, and mitochondrial dysfunction have been implicated in the pathogenesis and progression of PD. Essentially, damaged or dysfunctional mitochondrial system causes generation of reactive oxygen species (ROS), impaired energy (ATP) production, and stimulate pro-apoptotic signaling pathways, all of which influence the death of dopaminergic neurons in the SNpc [4]. The cells counteract these dyshomeostasis through a well-orchestrated cellular machinery called mitophagy. Mitophagy is a cytoprotective mechanism involved in the selective removal or degradation of damaged or worn-out mitochondrial within the cell through autophagy to maintain cellular or mitochondrial homeostasis [5]. Mitophagy relies especially on the PTEN-induced kinase 1 (PINK1) and Parkin, an E3 ubiquitin ligase functions. Reports have shown that PINK1 under physiological conditions exists in disintegrated form in a normal mitochondrial, however, during the depolarization of mitochondrial, PINK1 aggregate on the outer mitochondrial membrane surface to recruit Parkin, stimulating mitochondrial substrates ubiquitination through phosphorylation of both Parkin and ubiquitin, followed by autophagic degradation. Disruption of this signaling pathway through genetic mutation of PINK1 or PARK2 (encoding Parkin) has been implicated in the pathogenesis and progression of familial PD, reinforcing its significant neuroprotective role [6].
While familial PD is linked with inherited mutation and impaired PINK1/Parkin signaling-mediated defective mitophagic clearance, Sporadic PD has also been reported to manifest these pathological hallmarks which is also observed in neurotoxin experimental PD models [7]. These findings highlight the therapeutic implication of mitophagy restoration. The potential of some plant-derived phytochemicals in modulating cellular stress responses, mitochondrial homeostasis and autophagic alterations have been demonstrated. Importantly, phytonutrients including baicalein, berberine, quercetin, curcumin, and resveratrol have been reported to possess antioxidant, anti-inflammatory, signaling modulatory properties with capacity to stimulate mitochondrial quality function through PINK1/ Parkin pathway direct or indirect activation.
Providing insights into mechanisms by which these phytochemicals promote mitophagy will underscore development of promising mitochondria-directed nutriceuticals and adjuvant for the management of PD. This mechanistic narrative review therefore synthesizes data into the mechanisms of phytochemicals-mediated mitochondrial homeostasis restoration, modulation of PINK1/Parkindependent and -independent mitophagy, and dopaminergic neuroprotective mechanisms. This review also highlights important knowledge gaps by integrating evidence from molecular, biochemical, cellular, and animal studies as vehicle for the development of translational research aiding the development of phytochemical based nutraceuticals.
Methodology
A narrative review was conducted to synthesize mechanistic evidence of how phytochemicals promote mitophagy through the PINK1/Parkin pathway in experimental PD models. Databases including Google scholar and PubMed were searched and complemented by manual searches of references found in relevant literatures. Search criteria were confined to “Parkinson’s disease,” “dopaminergic neuroprotection”, “phytochemicals,” “PINK1”, “Parkin” and “mitophagy”, alongside combined concepts related to these words. Original articles focusing on the effects of naturally-derived phytochemical on mitochondrial homeostasis and mitophagy-associated signaling in vitro or in vivo PD models were emphasized. Essentially, articles were considered relevant to this review if they gave connection of molecular or biochemical mechanisms with PINK1/Parkin modulation, mitochondrial homeostasis, or promotion of neuronal integrity. Retrieved data was narratively structured into the following thematic sections; phytochemical classifications and natural sources, implicated molecular targets in mitophagy-associated pathways, phytochemicals effect on mitochondrial integrity, and the neuroprotective effects on the dopaminergic neurons. Studies’ findings were conceptually unified to underscore mechanistic signaling networks, pinpoint common signaling mechanisms, and propose future directions in favour of translational research.
Mitochondrial dysfunction and mitophagy in Parkinson’s disease
Pathogenesis and progression of PD is a multifactorial process with constellation of diverse environmental, biological, and genetic dynamics. Mainstream of PD till date are believed to be non-Mendelian and referred to as ‘idiopathic PD’ (IDP). The major huddle associated with IDP patients is the complex pathophysiology, and to date only partly understood. The Mendelian form of PD, termed “monogenic PD” (mPD) has been associated with several signaling pathways, the evaluation of which has enhanced our comprehension of the molecular basis of IDP [8]. Mechanistic investigations suggest that many of these signaling pathways converge on mitophagy, the main mitochondria quality control mechanism. This convergence makes dyshomeostasis in mitochondria a mechanistic bridge linking monogenic and idiopathic forms of PD. Mitochondrial dyshomeostasis has been linked to several known types of mPD, including pathways that are also pertinent to IDP. According to genome-wide association studies (GWAS), many genetic loci linked with IDP have been located within the genes associated with mPD. Mitochondrial dysfunction has been reported to be directly involved in many of these mPD genes, as well as the identified IDP risk loci [9]. At cellular level, mitochondrial dysfunction causes defective cellular clearance of impaired mitochondria, hyper-generation of reactive oxygen species, and sustained failure in biogenesis. According to Bose and Beal [10], mutations in PRKN, PINK1, and LRRK2 have been identified to cause distinct damages to mitochondrial-associated signaling pathways. Importantly, mutations in PRKN and PINK1 genes causes disruption in mitophagic labeling process and removal of depolarized mitochondrial, causing pathologic aggregation of the depolarized mitochondria. On the other way hand, pathogenic variant of LRK2 gene disrupts mitochondria trafficking and turnover by altering the activity of kinases. These disruptions together compromise the mitochondrial homeostasis in dopaminergic neurons. Although, dysfunctional cytoprotection against oxidative stress is directly related to DJ-1 PD, there is a plausible association with mitochondrial impairments, notwithstanding a lack of detail on mechanistic involvement [11]. Emerging evidence has shown that the loss of DJ-1 functions indirectly brings about exacerbation of mitochondria impairments increasing generation of reactive oxygen species, weakening cellular redox balance, and decreasing mitochondrial stress resistance. Accumulating studies demonstrate that these networks co-influence one another; every dysfunctional PD-associated gene product can directly or indirectly influence several pathways.
Role of mitochondrial impairment in the pathogenesis Parkinson’s disease
Mitochondrial impairment plays a pivotal role in the pathogenesis and progression of PD [12]. Several factors including neuronal energy failure, impairment of mitochondrial biogenesis, oxidative stress, variations to mitochondrial dynamics, calcium (Ca2+) dyshomeostasis, impaired mitophagy, and dysfunctional electron transport chain significantly contribute to dysfunctional mitochondrial induced PD [13, 14]. The mitochondrial abnormalities are not solo events but are mechanistically and intricately interconnected, with damaged mitophagy acting as a pivotal failure point in the clearance of mitochondrial that are dysfunctional. Aggregation of impaired mitochondria exacerbates oxidative stress and failure in biogenesis, thereby promoting vulnerability and death of dopaminergic neurons. Dysfunctional electron transport chain (complex Ⅰ) in the dopaminergic neurons of the SNpc causes failure in ATP production and oxidative stress. Damage to complex 1 causes leakage of electron and excessive generation of superoxide which directly impairs the integrity of mitochondria membrane and enzymatic action. Sustained oxidative stress further damages mitochondria signaling pathways required for energy sensing and quality control. Hyper-generation of reactive oxygen species in the SNpc impairs the mitochondrial proteins, DNA, and induces mitochondria lipid peroxidation, further potentiating the mitochondrial impairments. Inadequate endogenous and exogenous defensive mechanisms against mitochondrial-associated oxidative stress result into a vicious cycle of dysregulation leading to cellular impairment and ultimately dopaminergic cell death. This vicious cycle is strengthened by dysfunctional mitochondrial flux, which brings about limitations in the selective removal of severely impaired mitochondria. Resultantly, impairment of mitochondria becomes self-propagating within the dopaminergic neurons of the SNpc. Asides the major function of mitochondrial, which is ATP production, it has been documented that IPD and mPD are associated with mitochondrial-induced cellular dysfunction such as dysregulation of cell death via apoptosis, poor formation and export of iron-Sulphur (Fe-S) clusters, altered haem biosynthesis, Ca2+ dyshomeostasis, and altered control of cell division and growth [10]. Several of these cellular events are highly controlled and sustained mitochondrial homeostasis and turnover. Disruption of mitochondrial quality control and mitophagy, therefore associates failure in energy production with abnormal cell death signaling and metabolic dyshomeostasis in PD. Exposures to some environmental neurotoxins including MPTP and rotenone has been shown to induce these mitochondrial impairments, underpinning their involvements in the pathogenesis and progression of PD. These neurotoxins promote pathogenesis of PD primarily by inhibiting mitochondria complex I, thereby stressing endogenous antioxidant defense mechanisms and mitophagic capacity. Their effects further underpin impairments in mitochondrial function as a causal promoter rather than a secondary consequence of neuronal death or degeneration. Electrons can react with molecular oxygen after leaking from complex I and complex III of the electron transport chain during oxidative phosphorylation to form superoxide (O2− ), a reactive oxygen species (ROS) [15]. This process under physiological conditions is inhibited or occurs at a significantly reduced concentrations through the activities of mitochondrial antioxidants, including manganese superoxide dismutase (MnSOD) or SOD2), glutathione (GSH) and peroxiredoxins. Hydrogen peroxide is formed when MnSOD reacts with O2−, to remove H2O2 [16]. A reduction in the concentration or activity of GSH could be an onset and modifiable occurrence in the pathogenesis of PD in the SNpc. Depletion of glutathione as well as its activity amplifies mitochondria oxidative damage and indirectly disrupts the mitochondria quality control, mitophagy, which further amplifies dopaminergic neuronal death or degeneration.
Mitophagy as a dysfunctional mitochondrial clearance and quality-control mechanism
Mitophagy has been reported to play a key role in the pathophysiology of PD. Mitophagy removes damaged or dysfunctional mitochondrial from the SNpc through autophagy to maintain cellular function. This process is of particular importance to the physiology of neurons, where limited regenerative capacity requires a well-organized mitochondria quality control to maintain viability of the neuron for a long time. Two important genes, responsible for autosomal recessive early onset mPD are required to mediate mitophagy process; the PRKN and PINK1 genes [17]. The functionality of these genes is sequential and tightly regulated within a mitochondrial surveillance system. Alteration of this system disrupts the selective recognition and clearance of impaired mitochondria, thereby amplifying accumulation of mitochondria and neuronal stress. During normal physiological conditions, membrane potentials or healthy mitochondrial is sustained, preventing PINK1 aggregation on the outer mitochondria membrane surface. But during depolarization of the mitochondrial, PINK1 is stabilized on the surface causing E3 ubiquitin ligase Parkin recruitment from the cytosol [18].
Ubiquitin and Parkin are both phosphorylated by stabilized PINK1, thereby stimulating the activity of ParkinE3 ligase activity. There is an amplification of mitophagicsignaling pathway on account of the phosphorylationcascade, and ensure efficient targeting of impaired mitochondria for degradation. Numerous outer membraneproteins are further ubiquinated from the activated Parkincausing degradation of the dysfunctional mitochondrialthrough autophagosome-lysosome pathway. Recruitmentof autophagy receptors and adaptor proteins that connectimpaired mitochondria to the autophagosomal machineryis stimulated by ubiquitination of these substrates. Inhibition of this process causes an incomplete mitophagicflux and maintenance of dysfunctional mitochondria. Inkeeping with this, enhanced mitophagy has been reportedto be a therapeutic strategy as the process guarantees theremoval of damaged mitochondria, thus mitigating againstoxidative stress and bioenergetics failure [18]. Application of therapies that promote mitophagy therefore targetsa disease-modifying mechanism and not just downstreamsymptomatic consequences. High metabolic demand andincreased basal oxidative load of dopaminergic cells in theSNpc make them to be selectively vulnerable to impairedmitophagy, contributing to the progressive mitochondrialimpairment, synaptic dyshomeostasis, and neuronal death,the pathological hallmarks of PD [19]. Even a slightestdisruption in mitophagic process can efficiently cause adisproportionate effect on dopaminergic neuronal survival.
Recognition and clearance of dysfunctional mitochondria is disrupted through mutations of PINK1 or Parkin causing impaired mitochondrial aggregation-mediated oxidative stress. Accordingly, reduced mitophagic flux and increased numbers of swollen dysfunctional mitochondria are evidenced in postmortem studies of PD brains and cellular models [19]. These observations provide pathological evidences that dysfunctional mitophagy plays a critical role in mitochondrial dysfunction-induced PD.
The PINK1/Parkin pathway as a key regulator of mitophagy
Neural cells use specialized machinery, PINK1/Parkin pathway in clearing damaged or impaired mitochondria through mitophagy. Based on the status of membrane potential, this pathway acts as mitochondrial surveillance mechanism that identifies the damaged mitochondria from the healthy ones. PINK1 is transient serine/threonine kinase consisting of 581 amino acids [20]. Structurally, it has specialized domains including an N-terminal mitochondrial targeting sequence, a serine/threonine kinase region, a transmembrane domains, and a C-terminal autoregulatory region. Using these domains, PINK1 senses mitochondrial integrity and stimulates downstream signaling processes that is needed for the activation of mitophagy. The concentrations of PINK1 are low under physiological conditions through the proteolytic processing of mitochondria proteases that cleave and degrade PINK1 after translocation into healthy mitochondria, maintaining the cytosolic concentration of PINK1 at low levels [21].
An inappropriate activation of mitophagy is prevented by this constitutive degradation in mitochondria that are metabolically active. When the mitochondria membrane potential is polarized, a full length of PINK1 (63 kDa) is imported into the mitochondria by forming complexes with both outer and inner mitochondria membrane translocase [22]. This process converts the PINK1 into its active form (52 kDa) and then releases it into the cytosol for degradation by the ubiquitin-proteasome system. Translocation of PINK1 is stopped, causing increased concentrations of PINK1 on the outer mitochondrial membrane (OMM) - a key indicator of mitochondria depolarization and damage. Mitochondria depolarization is converted into a mitophagic signal through accumulation of PINK1 on the OMM, a molecular switch. Errors in cleavage processes and increased concentrations on PINK1 on the outer mitochondria membrane stabilizes PINK1 and becomes a downstream signal, including autophosphorylation of PINK1 and subsequent phosphorylation of cytosol, its inactive form of ubiquitin and the E3 ubiquitin ligase Parkin. Mitophagic signal is further amplified through phosphorylation of ubiquitin and Parkin by stimulating substrate identification and continuous activation of Parkin. This process is ensured by a feed-forward mechanism through an efficient labelling of impaired mitochondria for neuronal removal. Mitophagy mechanism is stimulated once PINK1 phosphorylates the Ub-like region of Parkin, activating it [22]. Mitochondria themselves are marked for degradation when active parkin ubiquitinates mitochondrial proteins such as Mfn1, VDAC1, and Mfn2. Recruitment of autophagy receptors that link impaired mitochondria to the autophagosomal machinery is stimulated by ubiquitination of these outer OMM. This complex process results into the PINK1/Parkin pathway [23]. Physiologically, this pathway is important as it preserves the neuronal-axonal mitochondrial homeostasis. The functionality of dopaminergic neurons depends on the maintenance of axonal mitochondrial quality, a phenomenon that depends on continuous transportation of mitochondria and localization of ATP biogenesis. This mechanism is achieved by local synthesis of PINK1 and transportation within the mitochondria, allowing mitophagic mechanism to be carried out in the axonal region of the neuron [23]. Figure 1 above shows the PINK1/Parkin-mediated mitophagy pathway in Parkinson’s disease
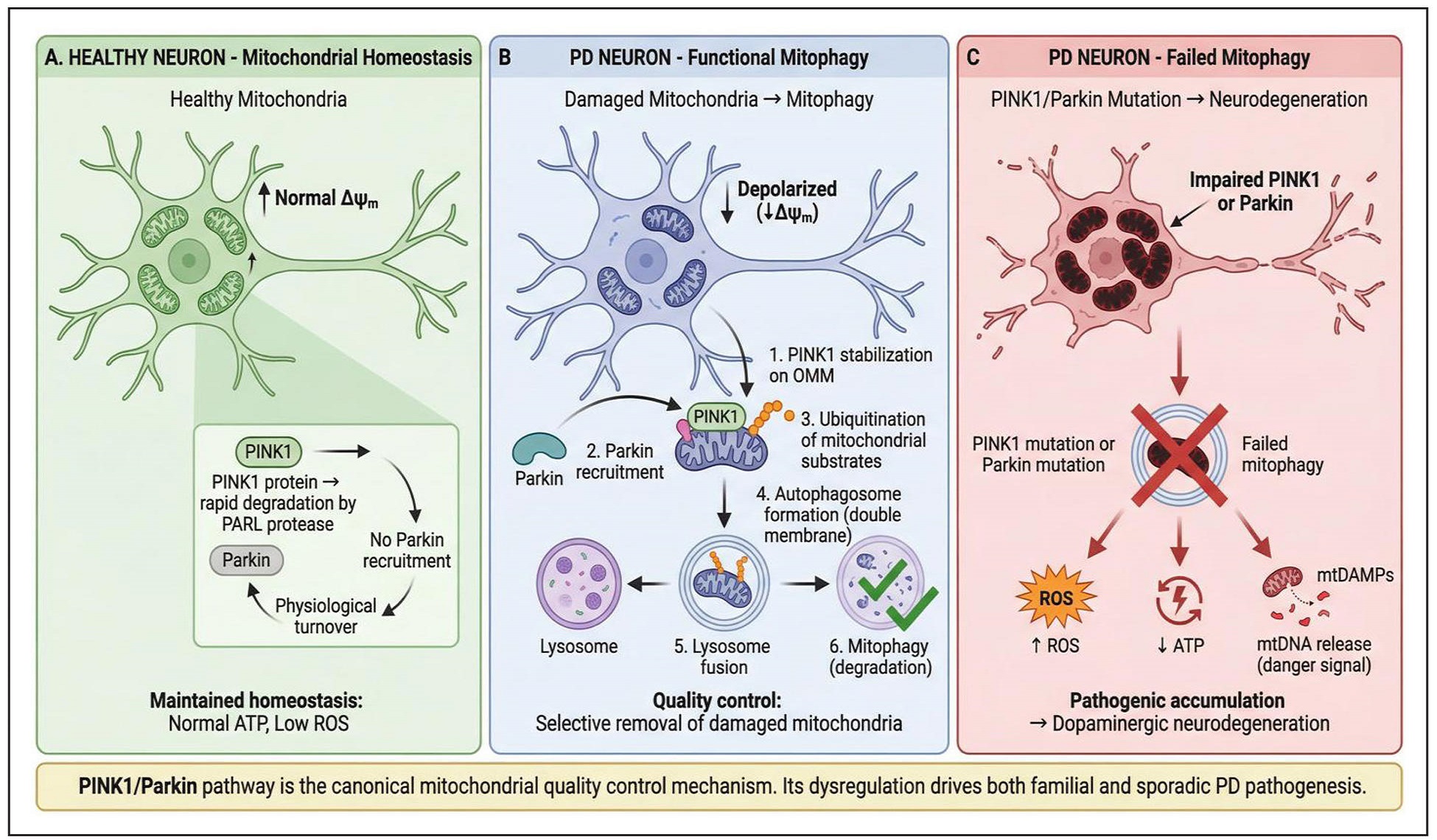
Figure 1. PINK1/Parkin-mediated mitophagy pathway in Parkinson’s disease. Three-panel comparison showing PINK1/Parkin-mediated mitophagy in healthy neurons (physiological turnover), PD neurons with functional mitophagy (quality control), and PD neurons with failed mitophagy (pathogenic accumulation leading to neurodegeneration). Establishes the biological foundation for understanding why mitophagy failure drives PD pathogenesis.
PINK1/Parkin pathway disruption and implica- tion in Parkinson’s disease
Death or loss of dopaminergic neurons in the SNpc is the pathological hallmarks of PD. This selective dopaminergic neuronal vulnerability is intricately associated with mitochondrial dysfunction and disrupts mitophagy, the mitochondrial quality control system. Mechanistically, mitochondrial overload and energy failures are associated with the progressive neurodegeneration with the SNpc [24]. The overload in mitochondria is a reflection of accumulation of impaired mitochondria produced by insufficient clearance through mitophagy. Dysfunctional energy biogenesis arising from dyshomeostasis compromises the synaptic transmission and inhibits neuronal integrity as well as survival signaling. While there is death of dopaminergic neurons, surviving neurons manifest Lewy bodies, the intracellular inclusion made up of aggregated alphasynuclein proteins. Overexpression of the parkin gene in the SNpc has been reported to mitigate the accumulation of alpha-synuclein [25]. This observation is suggestive of a physiologic interplay between mitochondrial quality and α-synuclein homeostasis. Parkin-stimulated mitophagy may limit the aggregation of α-synuclein indirectly by inhibiting or reducing mitochondria-mediated oxidative stress. Mutations of the gene encoding Parkin (PINK1 or PARK2) have been associated with the early onset of PD, with compromised homeostatic mechanism, causing defective recognition and clearance of impaired mitochondria. Inhibition of recognition process impairs mitophagic flux at the beginning. In effect, impaired mitochondria are sustained within the dopaminergic neurons and acts as a protracted source of cellular stress. This leads to accumulation of impaired mitochondria, causing generation of excess reactive oxygen species and hyper-generation of pro-apoptotic factors that potentiate dopaminergic death. Progressive oxidative damage and apoptotic signaling further potentiate dopaminergic neuronal degeneration or death via feed-forward mitochondria impairment. Collectively, mutations of PINK1 and parkin can work in consonance with other genetic mutations, resulting to exacerbated disease phenotypes [26]. This kind of genetic interaction underpins the intricate nature of mitochondria and mitophagy-associated networks in the pathogenesis of PD. A lot of IPD models have exhibited reduced activation of PINK1/Parkin in the absence of genetic mutation, reinforcing the assertion that environmental factors and age-associated oxidative stress can alternatively damage this pathway. Long-term oxidative stress and inflammatory signaling linked with aging may result into impairment of PINK1 stabilization or Parkin recruitment. This pathologic suppression of the pathway phenocopies geneticloss-of-function mutations. For instance, report has shown that individuals with biallelic PINK1/Parkin mutations manifest higher concentrations of cell-free circulating mitochondrial DNA (mtDNA) relative to age-linked PD [27]. Inflammation-associated mtDNA has been reported to be a biomarker for PINK1/Parkin-associated PD [27].
Mitochondrial DNA extrusion is an indication of defective mitochondria containment and further associates with impaired mitophagy and neuroinflammatory processes in PD.
Animal and cellular studies supporting PINK1/Parkin pathway disruption in the pathogenesis of PD
Studies have shown the pivotal role of PINK1/Parkin pathway in the maintenance of mitochondrial integrity.
This pathway acts like a focal mitochondrial quality control axis that involves integration of stress signaling with mitochondria removal from neurons. While there are limitations in some genetic-inclined PD models, they have contributed immensely to the understanding of PD pathogenesis and drug discovery. For instance, knockout of the PINK1 or Parkin genes in PD mouse model alone has given enough indication of Parkinsonism in mouse despite exhibition of impaired mitochondrial morphology and function, increased oxidative stress, and mild dopaminergic dysfunction [28]. These results reveal that the loss of PINK1 or Parkin genes is enough to bring about disruption in mitochondrial integrity even when there is no obvious neurodegeneration. Damage to the mitophagic signaling pathway therefore precedes and stimulates dopaminergic neurons to consequent degenerative insults. Of note is that even when these mice have genetic background with high concentrations of mitochondria mutations, it caused dopaminergic neuronal loss [28]. This event is an indication of a threshold effect whereby accumulation of mitochondria defects overburdens compensatory quality control mechanisms. Studies have shown that cGAS-stimulator of interferon genes (STING1), a component of cellular immune system mediates loss of dopaminergic neurons -deficient models of PD [29]. Disruption of mitophagic network stimulates the release of DNA from the mitochondria into the cytoplasm, potentiating CGAS-STING-dependent immune activation. This mechanism mediates a direct relationship between impairment in mitochondrial function and neuroinflammation in PD models. In the same vein a report has shown a correlation between plasma concentrations of free mitochondria DNA and the inflammation marker IL-6 patient with PINK-mutant and parkin-mutant PD [27]. Activation of STING proteins is required for the stimulation of inflammatory process in PINK1 knockout mice [30]. These findings point to the fact that damaged PINK1/Parkin signaling stimulates inflammation-induced neurodegeneration through mitochondria danger signaling.
The activity of PINK1/Parkin pathway has been reported to increase in PD. This significant increase is suggestive of a complimentary response to amplified mitochondria stress rather than persistent functional mitophagy. Essentially, this compensation may be inadequate with the progression of the disease. Importantly, mPD is associated with hyper-concentration of pS65-ubiquitin-positive structures, a biomarker of PINK1/Parkin activation in the SNpc. Surprisingly, pS65-ubiquitin has been reported to be absent in mature Lewy bodies but present in newly developed Lewy bodies and in cells that are devoid of these cytoplasmic inclusions [31]. This sequential distribution is suggestive of an early activation of PINK1/Parkin during the pathogenic processes and declines with maturation of the Lewy body. In keeping with this, PINK1/Parkin pathway is not only activated in mPD but also implicated in the pathogenesis of Sporadic PD. According to Gao et al. [32] knockout of USP30 gene stimulates mitophagy and offer neuroprotection against alpha-synuclein in mitochondrial dysfunction-induced PD. USP30 in this context acts as a negative regulator of mitophagy through inhibition of Parkin-induced ubiquitination. The mitophagy flux is therefore restored on account of this inhibition even in conditions where Parkin activity is compromised. This report asserts that potential therapeutic approach of USP30 in the restoration of mitophagy in the absence of parkin, the influencer of mitophagic mechanism. These findings underpin the importance of therapies targeting mitophagy regulator downstream or parallel to the PINK1/Parkin network.
Furthermore, report has shown structural alterations including mitochondria swelling, impaired cristae structure and flight muscle degeneration in drosophila models of PD lacking PINK1/Parkin [33]. At the ultrastructural level, such structural dysfunctions are suggestive of severe mitochondrial quality control inhibition. It has been demonstrated that these alterations can be mitigated through over expression of complementary gene, reinforcing their common pathway. Study have shown a reduction in mitophagic pathway, altered MMP, and increased vulnerability to neurotoxins such as MPTP and rotenone in human induced pluripotent stem cell (iPSC)-derived dopaminergic neurons carrying mutant PINK1 and parkin genes.
Reports from these human cellular models provide indications that damaged PINK1/Parkin signaling pathways directly disrupt the resilience of neuron to stress. PINK1/ Parkin network remains a pivotal regulatory mechanism or neuronal resilience and underpins its dyshomeostasis as critical pathogenic contributor in the pathogenesis and progression of PD [33]. Put together, reports from animals and cellular studies provide convergent evidence that neuroinflammation, mitochondrial dysfunction, and dopaminergic neurodegeneration in PD are directly mediated through disruption of PINK1/Parkin pathway.
Roles of phytochemicals in the modulation of mitophagic network
The roles of plant-derived bioactive components (phytochemicals) in regulating mitochondrial function have gained significant attention due to their antioxidant and anti-inflammatory properties. Studies have shown that plant derived bioactive components have capacity to activate mitophagic axis by modulating PINK1/Parkin pathway, thus promoting the clearance of impaired mitochondria and dopaminergic neurodegeneration. These phytochemicals are potential neuroprotective and therapeutic adjuncts in the management of neurodegenerative diseases including PD due to their capacity to enhance mitochondrial functions and reduce oxidative stress. The roles of resveratrol, baicalein, and berberine on mitochondrial functions are specifically highlighted in this review.
Resveratrol
Resveratrol is plant-derived phytochemical from peanuts, red wine, and red grapes associated with several biological, neuroprotective, and therapeutic properties [34], such as the activation of Sirtuin 1 (SIRT1) [35], a human homologue of yeast silent information regulator 2 (Sir2) protein [36]. Resveratrol mediates neuroprotective effects through its anti-inflammatory and anti-oxidant effects as reported in vitro and in vivo models of PD (Table 1), such as dopamine induced apoptosis in neuronal SH-SY5Y cells [37], 6OHDA-treated rat model of PD [38] and MPTP mice model of PD [39]. Previous report by Pallàs et al. [40] revealed neuroprotective action of resveratrol through activation of SIRT1, even though authors could not substantiate the exact mechanism of action. Lee et al. [41] also reported the possibility of impaired cellular organelles accumulation and impaired energy biogenesis due to SIRT1 inhibition, suggesting the capacity of SIRT1 to stimulate basal rate of autophagy. Wu et al. [42] revealed that resveratrol mediated its neuroprotective action via AMPK/SIRT1–autophagy axis. Mudo et al. [43] also reported the neuroprotective effects of resveratrol by acting on SIRT1/Peroxisome proliferator-activated receptorgamma coactivator-1α (PGC-1α) pathway. Authors produced transgenic mice that were overexpressing PGC1α in dopaminergic neurons and evaluated the degree of cell degeneration in the SNpc after treating the mice with MPTP. Results demonstrate that resveratrol treatment in this study significantly prevented dopaminergic neurons in vivo against MPTP [43].
Table 1.
Mechanistic studies describing the roles of resveratrol in the regulation of mitophagy-related signalling pathways in different models of Parkinson’s disease.
| Phytochemical | Experimental model | Main target pathway | Key outcome | Mitophagy/neuroprotective mechanism | Reference |
|---|---|---|---|---|---|
| Resveratrol | Rotenone-treated SH-SY5Y cells and α-synuclein–expressing PC12 cells | AMPK/SIRT1 signaling | ↑ markers of autophagy; ↓ apoptosis; ↓ accumulation of α-synuclein | Clearance of α-synuclein and neuronal survival promotion through activation of AMPK-induced SIRT1-dependent autophagy signaling, | [42] |
| MPTP-induced mice, SN4741 dopaminergic cells, and PGC1α transgenic models | SIRT1/PGC-1α axis | ↑ mitochondrial antioxidant enzymes; ↑dopamin- ergic neuronal survival | Activation of SIRT1 causes enhancement of PGC-1α through mitochondrial biogenesis and antioxidant defense | [43] | |
| rosophila melanogaster parkin mutant model | Antioxidant and mitochondrialprotection pathways | ↓ oxidative stress markers; ↑ survival and locomotor activity | Increase in the activity antioxidant enzyme and protection of mitochondrial integrity | [44] | |
| Fibroblasts from early-developed PD patients having mutations of Park2 gene | AMPK/SIRT1/PGC-1α signaling | ↑ ATP biogenesis; ↑ mitochondrial respiration ↓ oxidative stress | Promotion of mitochondrial metabolism through activation of mitochondrial biogenesis and autophagic clearance | [45] | |
| - Hydroxydopamine (6-OHDA)-induced rat model of Parkinson’s disease | PINK1/TRAP1 signaling and mitochondrial complex I regulation | ↑ mitochondrial membrane potential and complex I activity in SNpc; ↑ expression of MT-ND1 (complex I subunit) and PINK1; ↑ phosphorylation of TRAP1; ↓ cytochrome-c and apoptosis-inducing factor (AIF) release | Resveratrol liposome preserved the integrity of mitochondrial through activation of PINK1/TRAP1 signaling pathway, improving mitochondrial respiration and ATP biogenesis and reducing mitochondrial-induced apoptosis in SNpc dopaminergic neurons | [46] | |
| Human fibroblasts carryingmutated parkin gene | Ca²⁺–cAMP regulation and AMPK/ SIRT1/PGC-1α signalling | Restoration of Endoplasmic reticulum–mitochondria contact site function; increased mitochondrial homeostasis through modulation of Ca²⁺ and cAMP signaling | Resveratrol caused restoration of mitochondrial homeostasis by regulating endoplasmic reticulum–mitochondrial Ca²⁺ feedback and activating AMPK/SIRT1/PGC-1α signaling pathways | [47] | |
| PTP-induced mouse model of Parkinson’s disease | SIRT1-induced autophagy and α-synuclein clearance | ↑ SIRT1 activity; LC3 deacetylation; ↓ dopaminergic neuronal loss and behavioral deficits; and cytoplasmic redistribution promoting autophagic degradation of α-synuclein | Activation of SIRT1-dependent autophagy stimulates α-synuclein clearance and protects dopaminergic neurons | [48] |
Abolaji et al. [44] highlighted the potential of resveratrol to protect against parkin-mutant Parkinsonism in Drosophila melanogaster model. Authors observed that parkin-mutant mice exhibited locomotor dysfunction, increased generation of reactive oxygen species, and significant reduction in survival. These pathologies were inhibited through administration of resveratrol which increased the climbing function and survival of the mice via restoration of acetylcholinesterase activity and mitigation of dopaminergic cells against oxidative stress. Authors observed at molecular level that resveratrol increased the gene expressions of ple (tyrosine hydroxylase) and Sod1 (superoxide dismutase) against parkin-mutant mice [44].
In keeping with this, the mitochondria deficiency in mutant mice was restored in post resveratrol administration. A study conducted by Ferretta et al. [45] using fibroblast obtained parkin-mutant PD patients revealed significant mitochondrial dysfunction, associated with impaired respiration and impaired ATP biogenesis. Authors also observed that upon treatment with resveratrol, there was a significant activation of AMPK and SIRT1, the critical energy detectors that converge on PGC-1α, pivotal regulator of mitochondrial synthesis. The downregulated AMPK/ SIRT1/PGC-1α signaling pathway in parkin deficiency was restored. PGC-1α upregulated TFAM, SOD2, and cytochrome c genes, enhancing the antioxidant defense system and mitochondria biogenesis.
Protective role of resveratrol liposomes on dopaminergic cells has also been reported in 6-hydroxydopamine–in duced rat model of PD. The oral administration of resveratrol remarkably enhanced the activity of mitochondria complex I and increased its subunits MT-ND1, reinstating the homeostasis of electron transport chain in the SNpc. Mechanistically, resveratrol liposomes promoted the MMP, reduced the cytochrome c and apoptosis-inducing factors release, increased phosphorylated TRAP1 and the p-TRAP1/TRAP1 ratio, and upregulated the expression of PINK1 supporting mitophagy.
Perspectives from different experimental models of PD using resveratrol as an intervention highlight the emergence of resveratrol as one of the most significant plantderived phytochemicals capable of modulating mitochondrial quality control, mitophagy.
The mechanistic reviews on resveratrol collectively touch on the stimulation of the AMPK/SIRT1/PGC-1α pathway, the upstream modulator of PINK1/Parkin-dependent mitophagy pathway. Resveratrol restores the homeostasis of mitochondria through this signaling pathway, importantly through reduction of oxidative stress and prevention of dopaminergic cells in the SNpc from degeneration.
Resveratrol mechanistically acts by stimulating AMPK which in turn activates SIRT1-dependent deacetylation of LC3 and PGC-1α, the pivotal enzymes required for autophagy. This molecular crosstalk mediates the neuronal clearance of the hallmarks of PD pathology, impaired mitochondria and misfolded α-synuclein collections through autophagy. For instance in parkin-mutant fibroblast, resveratrol was also reported to control Ca²⁺ and cAMP homeostasis and increased the affinity of endoplasmic reticulum and mitochondria complex, suggesting a comprehensive intra-organelle communication and energy metabolism. These events reinforce the preservation of mitochondria dynamics, ATP biogenesis, and neuronal integrity within the SNpc. Put together, activation of SIRT1 plays a pivotal function of bridging metabolic regulation, autophagy, and mitochondrial homeostasis. Notably, SIRT1-influenced deacetylation and stimulation of downstream autophagy effectors and directly or indirectly potentiates the activation of PINK1/Parkin signaling pathway. In keeping with this, clearance of α-synuclein and restoration of MMP through mitophagy are all PINK1/Parkin signaling pathway-dependent.
Reports in Table 1 reinforce the assertion that resveratrol promote mitophagy with translational applicability in PD. Worthy of note is that resveratrol is pleiotropically functional yet coherent mechanistically, concurrently engaging AMPK (metabolic detector), SIRT1 (longevity proteins), and mitochondria control axis (PINK1/Parkin). This underpins the assertion that PD pathogenesis and progression is critically centered on the loss of dopaminergic neurons but a collapse of mitochondrial quality control mechanism, a pathology resveratrol experimentally inhibits. Moreover, the converging mechanism of action of resveratrol through a plethora of PD models confers on it an evolutionary mechanism of action, ranging from rodent models to cellular models. Resveratrol appears to display a disease modifying capability through coordination of biogenic restoration, inhibition of oxidative stress, and autophagy recycling rather than symptomatic relief, typical of some synthetic anti-PD drugs.
Berberine
Berberine is a plant-derived alkaloid associated with traditional use, importantly in Ayurvedic and Chinese medicine [49]. Plants such as Berberis, Coptis, and Phellodendron have been reported to be the natural sources of Berberine. There are numerous pharmacological potentials of berberine, making it gain significant attention in the medical research setting [50]. Specifically, amidst several health benefits of berberine, it stands out as potent anti-inflammatory, antidiabetic, and antioxidant agent [51]. Reports have also shown the role of berberine in maintaining mitochondrial function and mitophagy modulation [52]. Berberine’s mechanism of action is pleiotropic including direct activation of mitophagic pathway through the stimulation of AMPK, stimulation of hormetic cytoprotective at low concentration through PI3K/AKT, Nrf2/HO1, involved in pro-survival and antioxidant signaling, and restoration of mitochondrial impairment, including those associated with the deficiency of PINK1 (Table 2).
Table 2.
Summary of studies investigating roles of berberine in promoting dopaminergic neuroprotection and mitophagy-related mechanisms in
Parkinsonian models.
| Phytochemical | Experimental model | Main target pathway | Key outcome | Mitophagy/neuroprotective mechanism | Reference |
|---|---|---|---|---|---|
| Berberine | Human cell lines and PINK1-knockout mouse embryonic fibroblasts | AMPK-dependent mitophagy induction | ↑ mitophagy activation; ↑ ATP biogenesis ↑ mitochondrial respiration |
Activation of AMPK-mediated mitophagy homeostasis and compensatory mitochondrial biogenesis promoting mitochondrial quality control | [55] |
| 6-OHDA-treated PC12 cells and zebrafish model of Parkinson’s disease | PI3K/AKT and Nrf2/HO-1 signaling | ↑ dopaminergic neuronal survival at low doses; improved locomotor function | Promotion of neuronal survival through hormetic activation of antioxidant defense pathways | [54] | |
| 6-OHDA-treated SH-SY5Y dopaminergic cells | PI3K/Akt–Nrf2–HO-1 pathway | ↓ ROS generation; ↓ apoptosis; ↑ HO-1 expression |
Activation of Nrf2-mediated antioxidant signaling causes suppression of oxidative stress and promotes dopaminergic neuronal survival | [55] |
Um et al. [53] demonstrated the stimulatory effect of berberine on mitophagy. Authors revealed that berberine stimulated mitophagy across different human cell without distorting mitochondrial homeostasis or cellular integrity.
Remarkably, its mitophagy stimulatory effect was mediated through adenosine monophosphate (AMP)-activated protein kinase (AMPK), the canonical enzyme required for initiation of autophagy processes (ULK1 activation, mTOR suppression) which influences mitophagy induction and mitochondrial homeostasis. Authors noted that the activities of berberine does not resemble the (ULK1 activation, mTOR suppression), rather it acts by preserving baseline mitochondrial homeostasis while assisting in the removal of impaired mitochondria which eventually increase the production of ATP for cellular survival. A study conducted by Zhang et al. [54] using 6-Hydroxydopamine (6-OHDA)-induced PC12 cell model and zebrafish model of PD revealed that berberine displayed a biphasic (hormetic) dose response. Specifically, administration of low dose of berberine caused an increase in the viability of the cells and inhibited 6-OHDA-induced toxicity. Contrariwise, treatment with a high dose of berberine resulted to loss of cell viability and eventual cell loss. In this instance, berberine acted by increasing the expression of Bcl-2 through activation of the PI3K/ AKT signaling axis and also increases the expression of HO-1 through the activation of Nrf2 signaling cascade. The physiological implication of this is that PI3K/AKT activation promotes cellular integrity and survival and can associate with AKT/mTOR axis and autophagy modulator, while there is significant reduction in oxidative stress through Nrf2/HO-1, fostering mitochondria protection and promoting an efficient mitophagic cascade.
Dopaminergic neuroprotective effects of berberine are further asserted through a study conducted by Bae et al. [55] using 6-Hydroxydopamine (6-OHDA)-treated SHSY5Y dopaminergic neuronal cells. Authors revealed that 6-OHDA administration caused increase in the generation of reactive oxygen species in the SH-SY5Y cells. However, treatment with berberine at low dose significantly reduced the oxidative stress, inhibited caspase-3, and increased the expression of HO-1 through activation of Nrf2. Notably, Nrf2 knockout inhibited the expression of HO-1 and its protective role, reinforcing a direct causal association. In keeping with this, increased expression of Nrf2 is dependent on the upstream activation of PI3K/ Akt and p38 MAPK cascade signaling. Implicatively, the capacity of berberine to reverse mitochondria impairment in PINK1 knockout cells suggests its capacity in engaging mitophagy and ensuring mitochondrial quality control through PINK1-independent mechanisms including AMPK-induced mitophagy or promotion of mitochondrial biogenesis by activating BNIP3/NIX, FUNDC1 cascade signaling that compensate for impaired PINK1/Parkin signaling axis. The unique activation of mitophagy through a PINK1-independent axis makes berberine a little different from compounds that act solely through established ubiquitin-dependent mitophagy cascade.
Baicalein
Baicalein is a plant-derived flavonoid from Scutellaria baicalensis. Baicalein mediates inhibitory action on dopaminergic neuronal loss through activation of mitophagy via SIRT1/AMPK/mTOR signaling pathway. According to Chen et al. [56], administration of baicalein in 6-OHDA model of Parkinson’s disease caused reversal of mitochondria fragmentation, promoted autophagosome formation, and inhibited neuronal apoptosis. Baicalein restored the mitochondrial homeostasis through inhibition of miR-30b-5p, a microRNA that suppresses SIRT1 expression, in this manner; it relieves the inhibition imposed on the SIRT1 and stimulates the phosphorylation of AMPK. The mTOR, a negative regulator of autophagy is in turn suppressed on account of this activation, resulting to enhanced mitophagic process and improved mitochondrial biogenesis. Mechanistically, the action of baicalein is an indirect regulation of miRNA modulation and it is distinctively different compared to resveratrol which directly activates the SIRT1 through modulation of NAD+ , revealing a specialized upstream control mechanism over SIRT pathway. Importantly, both pathways converge on AMPK-mediated mTOR inhibition which positions them with the network of mitochondrial homeostatic mechanism preservation.
Curcumin
Report has also shown the capacity of an important polyphenolic compound, curcumin, derived from Curcuma longa, to selectively promote autophagic clearance of impaired mitochondria through upregulation on PINK1 and Parkin expression [57]. In the same vein, curcumin has been shown to promote the translocation of transcription factor EB (TFEB), a critical regulator of lysosomal biogenesis, which accelerates the disintegration of damaged cellular organelles by complementing the activities of mitophagy [58]. The mechanism of action of curcumin goes beyond just initiation of mitophagy, but it extends to the completion of the autophagy flux, a canonical mechanism that is impaired in the Parkinson’s disease. Curcumin links redox regulation to mitophagic homeostasis through activation of the Nrf2. The crosstalk between Nrf2 activation (oxidative defense) and PINK1/Parkin upregulation (mitochondria clearance) exceptionally positions curcumin as a good candidate having dual modulation of mitochondrial renewal, and having a striking difference compared with resveratrol which relies on the SIRT-LC3 deacetylation pathway [59].
Quercetin
Quercetin is a plant-derived flavonol reported to promote activation of AMPK and phosphorylation of ULK1, stimulating mitophagy independent of SIRT1 [60]. According to Lin et al. [61], administration of quercetin in MPP+-treated neuronal models caused preservation of mitochondria membrane potential and inhibited aggregation of α-synuclein. Remarkably, administration of quercetin also upregulated the expression of PINK1, indicating partial convergence with established PINK1/Parkin pathway. Put together, quercetin stimulates AMPK through direct energy sensing axis, showing a SIRT-independent mechanism unlike other compounds that appear to primarily activate AMPK via SIRT-mediated NAD+ modulation [62].
The mechanistic actions of individual phytochemicals discussed in this section provides insights into the molecular entry points into mitophagy regulation. These details are provided in Table 3, giving a comparative synthesis summarizing mitophagic-related outcomes across studies. In the same vein, Figure 2 provides the phytochemical enhancement of mitophagy. Figure 3 above shows how the phytochemical enhancement of mitophagy translates to dopaminergic neuroprotection through restoration of mitochondrial homeostasis.
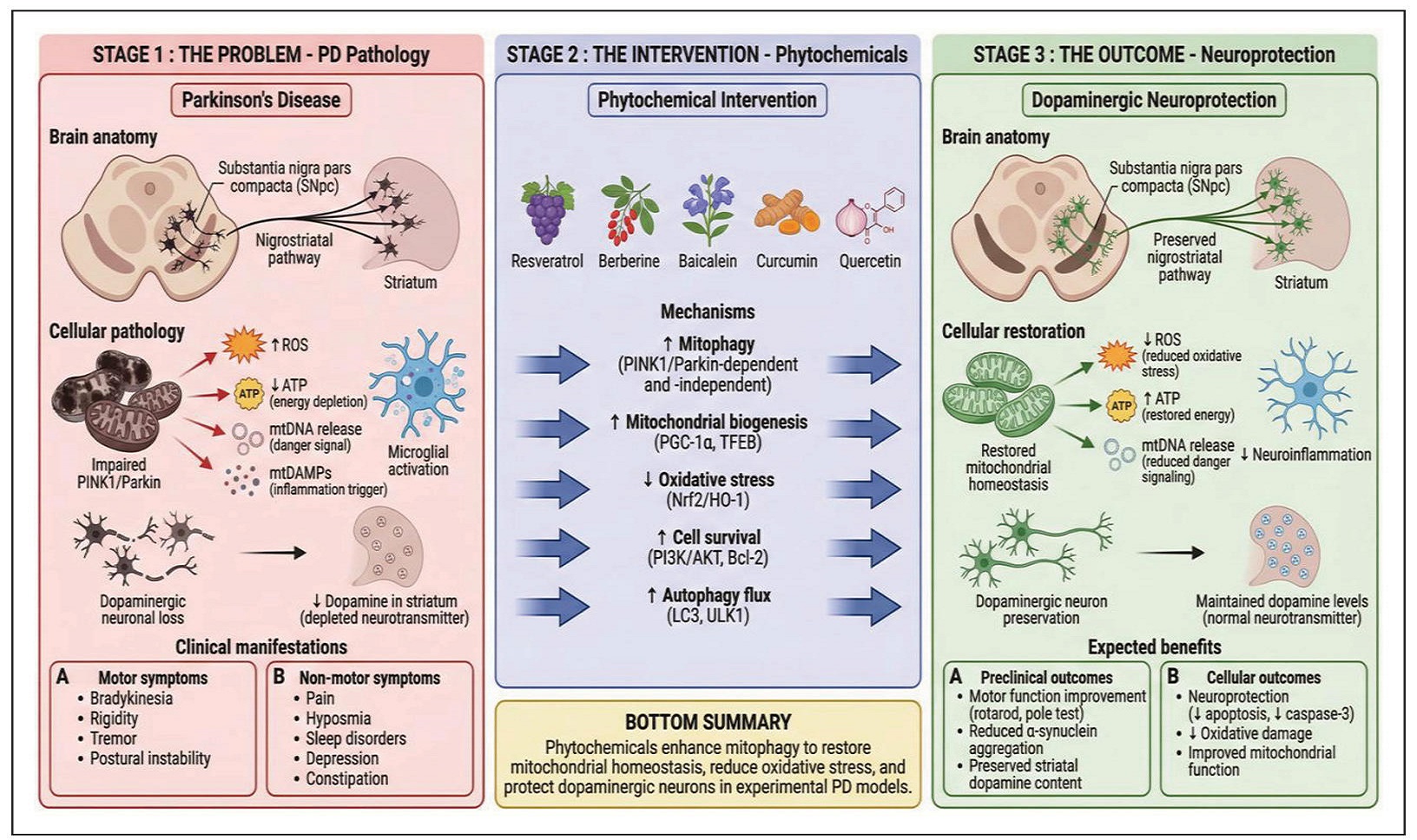
Figure 3. From mitophagy to dopaminergic neuroprotection: iranslational pathway. Left-to-right flow diagram showing the translational pathway from PD pathology (damaged mitochondrial, neurodegeneration, motor/non-motor symptoms) through phytochemical intervention (enhanced mitophagy via multiple mechanisms) to neuroprotection (restored mitochondrial homeostasis, preserved dopaminergic neurons, motor function improvement)
Table 3.
Mechanistic studies on the roles of quercetin and curcumin in the enhancement of mitophagy in Parkinson’s disease.
| Phytochemical | Model used | Main mechanism | Key outcome | Proposed mechanism | Reference |
|---|---|---|---|---|---|
| Quercetin | 6-OHDA rat & PC12 cells | AMPK–ULK1 activation | ↑ mitophagy markers; ↓ neuronal loss |
PINK1-associated mitochondrial quality control | [62] |
| Curcumin | MPTP & 6-OHDA models | Nrf2 activation; JNK inhibition | ↑ mitochondrial stability; ↓ inflammation |
Enhanced mitophagic flux & neuronal survival | [59, 63] |
Integration of mechanistic strategies of phyto- chemical-modulated mitophagy in PD
The loss of dopaminergic neurons in PD is intricately linked to dyshomeostasis in mitochondrial function and ultimate failure of mitophagy, the main mitochondrial quality control mechanism. While the above reviewed phytochemicals are structurally and pharmacologically different, there appears a focal feature which centers on mitochondrial homeostasis, regulating mitophagic function. These phytochemicals modulate interrelated signaling processes that control the integrity of mitochondria, oxidative balance, and viability of dopaminergic neurons of the SNpc rather than acting through individual targets.
PINK1/Parkin signaling cascade, the key regulator of mitophagy operates at the core of this convergence. Exposure to toxins induces neuroinflammation and mitochondria depolarization, a shared consequence of oxidative stress. This inhibits import and proteolytic degradation of PINK1, resulting in outer mitochondria membrane accumulation of PINK1. Stabilized PINK1 is autophosphorylated which in effect activates the phosphorylation of ubiquitin and Parkin, thereby activating Parkin’s E3 ubiquitin ligase function. Important outer mitochondria membrane proteins are ubiquitinated by the activated Parkin. This process characterizes the impaired mitochondria for recognition by autophagy receptors and consequential removal through the autophagosome-lysosome axis.
Preservation or promotion of this signaling pathway is an important focus on which many phytochemicals perform their modulatory functions in the dopaminergic neurons.
Persistence of dysfunctional mitochondria in the dopaminergic neurons is limited through phytochemical inducedmodulation of PINK1/Parkin pathway, which facilitates an efficient mitophagic flux. This process enhances energy biogenesis through reduction in mitochondria-derived oxygen species and inhibits the release of pro-inflammatory mitochondria components, including mitochondrial DNA. Inhibition of this downstream stress signals mitigates activation of inflammation-related networks, such as cGAS– STING signaling, bringing about reduction in the amplification of neuroinflammation that stimulates progressive neuronal loss in PD.
Emerging evidence indicates that phytochemical-induced reinforcement of mitophagy particularly enhances the viability and survival of SNpc dopaminergic neurons, the high metabolic demanding neurons with increased basal oxidative load and low resistance to mitochondria stress. In SNpc dopaminergic neuron, even a low mitochondrial dysfunction can cause cumulative neuronal damage, synaptic dysfunction, and eventual neuronal death. Application of phytochemicals may confer selective dopaminergic neuroprotection by stabilizing mitophagic process, the mitochondrial quality control system.
The highlighted observations collectively reinforce a network-based view wherein phytochemicals function by modulating the mitophagy, the mitochondrial quality control system rather than as a single-target agent. The ability of these phytochemicals to stimulate upstream mitochondria stability, amplify PINK1/Parkin-dependent mitophagy, and inhibit downstream oxidative and inflammatory signaling pathways suggests a mechanistic pathway linking the application of phytochemicals as neuroprotectants for dopaminergic neurons. This consolidative perspective gives indications that mitophagy plays a pivotal role in the application of therapies in PD and underpins the potential of phytochemical approaches in restoring the resilience of mitochondria in neurodegenerative conditions, especially PD.
Limitations
Despite the robust preclinical data, there are still limitations as most conclusions are drawn from cell culture and rodent models. There are few translational studies or pharmacokinetic validations in human subjects. Of importance is the suboptimal bioavailability and limited or inability of some phytochemicals to cross the blood-brain barrier, raising arguments about whether the central nervous system can receive the required concentration needed for its protection. Another striking limitation to some these preclinical studies is the therapeutic optimization which is challenged by streamlined dose windows and hormetic responses as displayed by most phytochemicals where benefits obtained from low dose may not scale predictably to human physiology and therefore necessitate a thorough pharmacokinetics and pharmacodynamics characterization. Mechanistically, AMPK activation is a canonical mechanism of most phytochemicals; however, the body of knowledge is still limited about the specific downstream mitophagy pathways involved by different phytochemicals. Furthermore, potential questions about metabolic or potential off-target effect of prolonged AMPK activation need to be put into consideration.
It is important to acknowledge that the current body of knowledge about phytochemical-mediated mitophagy in PD is predominantly based on in vitro experiments and animal models, which may not totally provide the intricate pathophysiology and progression of PD as experienced by humans. Notably, variations in dosing regimen, experimental designs, and PD models across studies make direct comparability of findings difficult. Notwithstanding the robustness of many preclinical data, providing compelling evidence for the neuroprotective potential of phytochemicals directed to mitophagic pathways, well-designed translationally and clinical investigations are further needed in order to determine the therapeutic effectiveness of phytochemicals in individuals with PD.
Translational and therapeutic implications
The collective evidence reviews are clear indications that phytochemical-mediated mitophagic modulation represents a potential paradigm shift from purely symptomatic dopamine replacement therapy towards mitochondrial quality-control-based dopaminergic neuroprotection. Phytochemicals may serve as adjuvant therapeutic candidates, with capacity to enhance neuronal survival by restoring mitochondrial integrity rather than functioning as a direct dopaminergic substitute. On this premise, potential preclinical effects of the phytochemicals can translate into meaningful clinical outcomes.
Integration of phytochemicals as adjunctive strategies alongside existing therapeutic approaches, such as LDOPA therapy and neurorehabilitation interventions, may offer synergistic benefits by addressing the pathophysiological mechanisms as well as symptomatic deficits associated with PD. Despite promising preclinical evidence, bioavailability challenges can be overcome through advances in intranasal delivery systems, nanoformulation, and structural optimization, which may further increase the bioavailability of the compounds in the CNS, thereby improving translational feasibility. In the same vein, future clinical trials should incorporate mitophagy biomarkers (mtDNA, LC3-II, PINK1 in CSF/blood) to assess target engagement. Figure 4 above shows the limitations and the solutions to bridging the knowledge gaps in the development of evidenced-based, mitochondria-directed nutraceuticals for PD management.
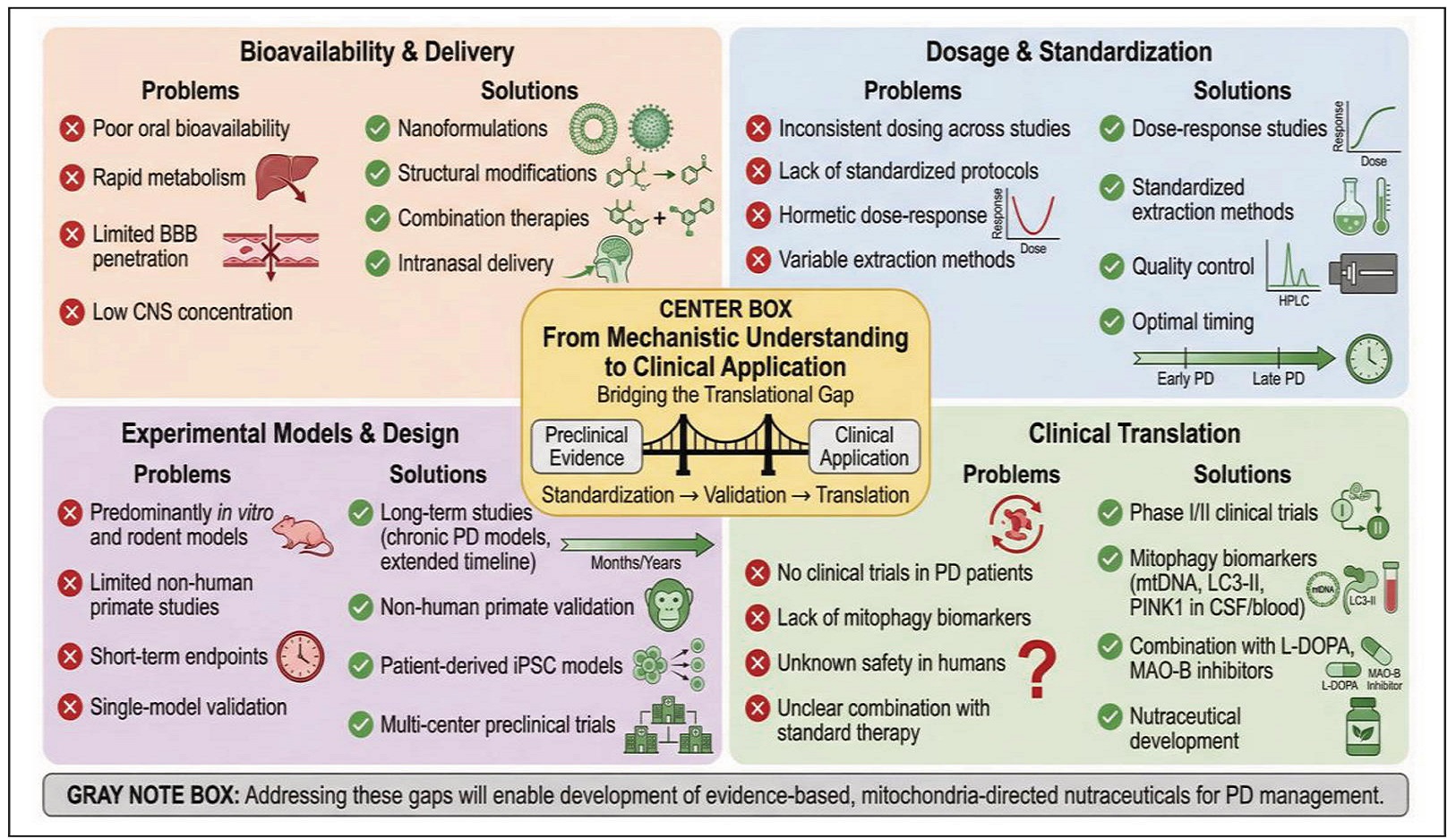
Figure 4. Knowledge gaps and future directions: bridging the translational gap. Four-quadrant diagram addressing critical limitations (bioavailability, dosage / standardization, experimental design, clinical translation) and proposing solutions (nanoformulations, dose-response studies, long-term / primate models, clinical trials with biomarkers) for translating phytochemical-enhanced mitophagy from preclinical evidence to clinical application.
Declarations
Modulation of the PINK1/Parkin signaling pathways remains a pivotal mechanism in the restoration of mitochondrial homeostasis and enhancement of mitophagy in Parkinson’s disease. Application of phytochemicals, which are plant-derived molecules, has shown promising effects in driving mitochondrial homeostasis. Reports from molecular and cellular studies using animal models of Parkinson’s disease reveal that phytochemicals such as resveratrol, berberine, and baicalein offer dopaminergic neuroprotective property by stabilizing PINK1, promoting Parkin translocation, improving mitochondria membrane potential, and stimulating biological factors including AMPK, SIRT1, Nrf2, and PGC-1α. Clearance of impaired mitochondria is enhanced through these mechanisms, which inhibit generation of reactive oxygen species, and protect the integrity of dopaminergic cells.
There are still limitations, specifically knowledge gaps despite the up to date stimulating findings. Importantly, there is poor body of knowledge about bioavailability, optimal dosing, pharmacokinetics, and long term safety of phytochemical interventions. Many studies are focused on preclinical research which is limited by heterogeneity in experimental design, limited mechanistic depth, and a poor standardized outcome evaluation. More rigorous mechanistic researches are required to bridge this knowledge gap and harmonize methodologies to promote welldesigned translational studies involving human-obtained cellular systems and early phase of clinical trials.
A better understanding of the mechanisms by which phytochemicals modulate PINK1/Parkin-dependent and independent mitophagy give a critical framework for the development of mitochondria-focused adjunctive and nutraceutical therapies. Mitophagic axis remains a disease modifying approach, and there is need to investigate on targeting it as a potential strategy in Parkinson’s disease management. Further investigation of the molecular associations remains critical for promoting phytochemical based strategies from preclinical models to clinical strategy.
Declarations
Author contributions
Patrick Oluwole Abolarin: data curation, methodology, writing–original draft, writing–review and editing; Peace Ayo Olaoluwa: review and editing; Ayodeji Johnson Ajibare: methodology, writing, figure design, review and editing; Abraham Olufemi Asuku: methodology, writing, figure review and editing; Gideon Opeyemi Ayilara: review and editing.
Financial support and sponsorship
None.
Conflicts of interest
Not applicable.
References
1. Abolarin P, Amin A, Nafiu A, Ogundele O, & Owoyele B. Optimization of Parkinson’s disease therapy with plant extracts and nutrition’s evolving roles. IBRO Neuroscience Reports, 2024, 17: 1-12. [Crossref]
2. Asuku A, Ayinla M, Olajide T, Ajagun E, Ganiyu K, Hamza Y, et al (2024). Diagnosis and biomarkers of Parkinson’s disease. in: zebrafish as a model for Parkinson’s Disease, Taylor and Francis, CRC Press. [Crossref]
3. Ajagun E, Asuku A, Ayinla M, Abdulsalam T, Ajibare A, Olajide T, et al (2024). Pharmacotherapy of Parkinson’s disease and their limitations. In: zebrafish as a model for Parkinson’s disease, Taylor and Francis, CRC Press. [Crossref]
4. Picca A, Guerra F, Calvani R, Romano R, Coelho-Júnior H, Bucci C, et al. Mitochondrial dysfunction, protein misfolding and neuroinflammation in Parkinson’s disease: roads to biomarker discovery. Biomolecules, 2021, 11(10): 1508-1519. [Crossref]
5. Wang W, Han R, He H, Li J, Chen S, Gu Y, et al. Administration of quercetin improves mitochondria quality control and protects the neurons in 6-OHDA-lesioned Parkinson’s disease models. Aging, 2021, 13(8): 11738-11751. [Crossref]
6. Asuku A, Ogungbangbe G, & Fajemidagba G. Gene therapy for Parkinson’s disease: a new frontier in neurodegenerative diseases. Exploration of Neuroprotective Therapy, 2025, 5: 1004119. [Crossref]
7. Sarkar A, Rasheed M, & Singh M. Redox modulation of mitochondrial proteins in the neurotoxicant models of Parkinson’s disease. Antioxid Redox Signal, 2023, 38(1012): 824-852. [Crossref]
8. Hernandez D, Reed X, & Singleton A. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J Neurochem, 2016, 139 Suppl 1: 59-74. [Crossref]
9. Lucchesi M, Biso L, Bonaso M, Longoni B, Buchignani B, Battini R, et al. Mitochondrial dysfunction in genetic and non-genetic Parkinson ’s disease. Int J Mol Sci, 2025, 26(9): 4451-4463. [Crossref]
10. Bose A, & Beal M. Mitochondrial dysfunction in Parkinson’s disease. J Neurochem, 2016, 139 Suppl 1: 216-231. [Crossref]
11. Dolgacheva L, Berezhnov A, Fedotova E, Zinchenko V, & Abramov A. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J Bioenerg Biomembr, 2019, 51(3): 175-188. [Crossref]
12. Golpich M, Amini E, Mohamed Z, Azman Ali R, Mohamed Ibrahim N, & Ahmadiani A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: pathogenesis and treatment. CNS Neurosci Ther, 2017, 23(1): 5-22. [Crossref]
13. Park J, Davis R, & Sue C. Mitochondrial dysfunction in Parkinson’s disease: new mechanistic insights and therapeutic perspectives. Curr Neurol Neurosci Rep, 2018, 18(5): 21-33. [Crossref]
14. Grünewald A, Kumar K, & Sue C. New insights into the complex role of mitochondria in Parkinson’s disease. Prog Neurobiol, 2019, 177: 73-93. [Crossref]
15. Rani L, & Mondal A. Emerging concepts of mitochondrial dysfunction in Parkinson’s disease progression: pathogenic and therapeutic implications. Mitochondrion, 2020, 50: 25-34. [Crossref]
16. Mischley L, Conley K, Shankland E, Kavanagh T, Rosenfeld M, Duda J, et al. Central nervous system uptake of intranasal glutathione in Parkinson’s disease. NPJ Parkinsons Dis, 2016, 2: 16002. [Crossref]
17. Pickrell A, & Youle R. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron, 2015, 85(2): 257-273. [Crossref]
18. Truban D, Hou X, Caulfield T, Fiesel F, & Springer W. PINK1, Parkin, and mitochondrial quality control: what can we learn about Parkinson’s disease pathobiology? J Parkinsons Dis, 2017, 7(1): 13-29. [Crossref]
19. Ni A, & Ernst C. Evidence that substantia nigra pars compacta dopaminergic neurons are selectively vulnerable to oxidative stress because they are highly metabolically active. Front Cell Neurosci, 2022, 16: 826193. [Crossref]
20. Sever T, & Erbas O. The role of the PINK1-Parkin pathway in mitophagy and its implications in neurodegenerative disorders. Journal of Experimental and Basic Medical Sciences, 2025, 5: 264-270. [Crossref]
21. Rüb C, Wilkening A, & Voos W. Mitochondrial quality control by the Pink1/Parkin system. Cell Tissue Res, 2017, 367(1): 111-123. [Crossref]
22. Zorova L, Popkov V, Plotnikov E, Silachev D, Pevzner I, Jankauskas S, et al. Mitochondrial membrane potential. Analytical Biochemistry, 2018, 552: 50-59. [Crossref]
23. McLelland G, Goiran T, Yi W, Dorval G, Chen C, Lauinger N, et al. Mfn2 ubiquitination by PINK1/Parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. Elife, 2018, 7: e32866. [Crossref]
24. Woo J, Cho H, Seol Y, Kim S, Park C, Yousefian-Jazi A, et al. Power failure of mitochondria and oxidative stress in neurodegeneration and its computational models. Antioxidants, 2021, 10(2): 229-236. [Crossref]
25. Yasuda T, & Mochizuki H. The regulatory role of α-synuclein and parkin in neuronal cell apoptosis; possible implications for the pathogenesis of Parkinson’s disease. Apoptosis, 2010, 15(11): 1312-1321. [Crossref]
26. Nuytemans K, Theuns J, Cruts M, & Van Broeckhoven C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat, 2010, 31(7): 763-780. [Crossref]
27. Borsche M, König I, Delcambre S, Petrucci S, Balck A, Brüggemann N, et al. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain, 2020, 143(10): 3041-3051. [Crossref]
28. Pinto M, Nissanka N, & Moraes C. Lack of Parkin anticipates the phenotype and affects mitochondrial morphology and mtDNA levels in a mouse model of Parkinson’s disease. J Neurosci, 2018, 38(4): 1042-1053. [Crossref]
29. Gąssowska-Dobrowolska M, Olech-Kochańczyk G, Culmsee C, & Adamczyk A. Novel insights into Parkin–mediated mitochondrial dysfunction and “mito-inflammation” in α-synuclein toxicity. The role of the cGAS–STING signalling pathway. J Inflamm Res, 2024, 17: 4549-4574. [Crossref]
30. Standaert D, & Childers G. Alpha-synuclein–mediated DNA damage, STING activation, and neuroinflammation in Parkinson’s disease. Proceedings of the National Academy of Sciences, 2022, 119(17): e2204058119. [Crossref]
31. Barodia S, McMeekin L, Creed R, Quinones E, Cowell R, & Goldberg M. PINK1 phosphorylates ubiquitin predominantly in astrocytes. NPJ Parkinsons Dis, 2019, 5: 29-39. [Crossref]
32. Gao X, Yang T, Gu Y, & Sun X. Mitochondrial dysfunction in Parkinson’s disease: from mechanistic insights to therapy. Front Aging Neurosci, 2022, 14: 885500. [Crossref]
33. Guo M. Drosophila as a model to study mitochondrial dysfunction in Parkinson’s disease. Cold Spring Harb Perspect Med, 2012, 2(11): a009944. [Crossref]
34. Kung H, Lin K, Kung C, & Lin T. Oxidative stress, mitochondrial dysfunction, and neuroprotection of polyphenols with respect to resveratrol in Parkinson’s disease. Biomedicines, 2021, 9(8): 918-925. [Crossref]
35. Albani D, Polito L, Batelli S, De Mauro S, Fracasso C, Martelli G, et al. The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1-42) peptide. J Neurochem, 2009, 110(5): 1445-1456. [Crossref]
36. Haigis M, & Guarente L. Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes Dev, 2006, 20(21): 2913-2921. [Crossref]
37. Lee M, Kang S, Poncz M, Song K, & Park K. Resveratrol protects SH-SY5Y neuroblastoma cells from apoptosis induced by dopamine. Exp Mol Med, 2007, 39(3): 376-384. [Crossref]
38. Jin F, Wu Q, Lu Y, Gong Q, & Shi J. Neuroprotective effect of resveratrol on 6-OHDA-induced Parkinson’s disease in rats. Eur J Pharmacol, 2008, 600(1-3): 78-82. [Crossref]
39. Blanchet J, Longpré F, Bureau G, Morissette M, DiPaolo T, Bronchti G, et al. Resveratrol, a red wine polyphenol, protects dopaminergic neurons in MPTP-treated mice. Progress in neuro-psychopharmacology and biological psychiatry, 2008, 32(5): 1243-1250. [Crossref]
40. Pallàs M, Casadesús G, Smith M, Coto-Montes A, Pelegri C, Vilaplana J, et al. Resveratrol and neurodegenerative diseases: activation of SIRT1 as the potential pathway towards neuroprotection. Curr Neurovasc Res, 2009, 6(1): 70-81. [Crossref]
41. Lee I, Cao L, Mostoslavsky R, Lombard D, Liu J, Bruns N, et al. A role for the NAD-dependent deacetylase SIRT1 in the regulation of autophagy. Proc Natl Acad Sci USA, 2008, 105(9): 3374-3379. [Crossref]
42. Wu Y, Li X, Zhu J, Xie W, Le W, Fan Z, et al. Resveratrolactivated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals, 2011, 19(3): 163-174. [Crossref]
43. Mudò G, Mäkelä J, Di Liberto V, Tselykh T, Olivieri M, Piepponen P, et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell Mol Life Sci, 2012, 69(7): 1153-1165. [Crossref]
44. Abolaji A, Adedara A, Adie M, Vicente-Crespo M, & Farombi E. Resveratrol prolongs lifespan and improves 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced oxidative damage and behavioural deficits in Drosophila melanogaster. Biochem Biophys Res Commun, 2018, 503(2): 1042-1048. [Crossref]
45. Ferretta A, Gaballo A, Tanzarella P, Piccoli C, Capitanio N, Nico B, et al. Effect of resveratrol on mitochondrial function: implications in parkin-associated familiar Parkinson’s disease. Biochim Biophys Acta, 2014, 1842(7): 902915. [Crossref]
46. Chen J, Liu Q, Wang Y, Guo Y, Xu X, Huang P, et al. Protective effects of resveratrol liposomes on mitochondria in substantia nigra cells of parkinsonized rats. Ann Palliat Med, 2021, 10(3): 2458-2468. [Crossref]
47. Signorile A, Ferretta A, Pacelli C, Capitanio N, Tanzarella P, Matrella M, et al. Resveratrol treatment in human parkin-mutant fibroblasts modulates cAMP and calcium homeostasis regulating the expression of mitochondriaassociated membranes resident proteins. Biomolecules, 2021, 11(10): 1511-1526. [Crossref]
48. Guo Y, Dong S, Cui X, Feng Y, Liu T, Yin M, et al. Resveratrol alleviates MPTP-induced motor impairments and pathological changes by autophagic degradation of α-synuclein via SIRT1-deacetylated LC3. Mol Nutr Food Res, 2016, 60(10): 2161-2175. [Crossref]
49. Habtemariam S. Recent advances in berberine inspired anticancer approaches: from drug combination to novel formulation technology and derivatization. Molecules, 2020, 25(6): 1426-1437.
50. Song D, Hao J, & Fan D. Biological properties and clinical applications of berberine. Front Med, 2020, 14(5): 564-582. [Crossref]
51. Gasmi A, Asghar F, Zafar S, Oliinyk P, Khavrona O, Lysiuk R, et al. Berberine: pharmacological features in health, disease and aging. Curr Med Chem, 2024, 31(10): 1214-1234. [Crossref]
52. Fang X, Wu H, Wei J, Miao R, Zhang Y, & Tian J. Research progress on the pharmacological effects of berberine targeting mitochondria. Front Endocrinol, 2022, 13: 982145. [Crossref]
53. Um J, Lee K, Kim Y, Lee D, Kim E, Kim D, et al. Berberine induces mitophagy through adenosine monophosphateactivated protein kinase and ameliorates mitochondrial dysfunction in PINK1 knockout mouse embryonic fibroblasts. Int J Mol Sci, 2023, 25(1): 219-228. [Crossref]
54. Zhang C, Li C, Chen S, Li Z, Jia X, Wang K, et al. Berberine protects against 6-OHDA-induced neurotoxicity in PC12 cells and zebrafish through hormetic mechanisms involving PI3K/AKT/Bcl-2 and Nrf2/HO-1 pathways. Redox Biol, 2017, 11: 1-11. [Crossref]
55. Bae J, Lee D, Kim Y, Gil M, Lee J, & Lee K. Berberine protects 6-hydroxydopamine-induced human dopaminergic neuronal cell death through the induction of heme oxygenase-1. Mol Cells, 2013, 35(2): 151-157. [Crossref]
56. Chen M, Peng L, Gong P, Zheng X, Sun T, Zhang X, et al. Baicalein induces mitochondrial autophagy to prevent Parkinson’s disease in rats via miR-30b and the SIRT1/ AMPK/mTOR pathway. Front Neurol, 2021, 12: 646817. [Crossref]
57. Jin Z, Chang B, Wei Y, Yang Y, Zhang H, Liu J, et al. Curcumin exerts chondroprotective effects against osteoarthritis by promoting AMPK/PINK1/Parkin-mediated mitophagy. Biomedicine & Pharmacotherapy, 2022, 151: 113092. [Crossref]
58. Radbakhsh S, Kesharwani P, & Sahebkar A. Therapeutic potential of curcumin in autophagy modulation: insights into the role of transcription factor EB. Mutation Research - Fundamental and Molecular Mechanisms of Mutagenesis, 2024, 829: 111879. [Crossref]
59. Pan J, Li H, Ma J, Tan Y, Xiao Q, Ding J, et al. Curcumin inhibition of JNKs prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease through suppressing mitochondria dysfunction. Transl Neurodegener, 2012, 1(1): 16-28. [Crossref]
60. Gravandi M, Fakhri S, Zarneshan S, Yarmohammadi A, & Khan H. Flavonoids modulate AMPK/PGC-1α and interconnected pathways toward potential neuroprotective activities. Metab Brain Dis, 2021, 36(7): 1501-1521. [Crossref]
61. Lin Z, Liu Y, Xue N, Zheng R, Yan Y, Wang Z, et al. Quercetin protects against MPP+/MPTP-induced dopaminergic neuron death in Parkinson’s disease by inhibiting ferroptosis. Oxid Med Cell Longev, 2022, 2022: 7769355. [Crossref]
62. Wang S, Long H, Hou L, Feng B, Ma Z, Wu Y, et al. The mitophagy pathway and its implications in human diseases. Signal Transduct Target Ther, 2023, 8(1): 304-315. [Crossref]
63. Tripanichkul W, & Jaroensuppaperch E. Ameliorating effects of curcumin on 6-OHDA-induced dopaminergic denervation, glial response, and SOD1 reduction in the striatum of hemiparkinsonian mice. Eur Rev Med Pharmacol Sci, 2013, 17(10): 1360-1368.