Open Access | Research
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
Neuroprotective effect of aldehyde trapping agent, hydralazinein a Parkinson’s disease mouse model
* Corresponding author: Elizabeth Fernandez
Mailing address: Sam and Ann Barshop Institute for Longevity and Aging Studies, UT Health at San Antonio, 4939 Charles Katz Dr, San Antonio, TX, USA.
Email: fernandeze@uthscsa.edu
Received: 13 November 2025 / Revised: 19 November 2025 / Accepted: 11 March 2026 / Published: 31 March 2026
DOI: 10.31491/APT.2026.03.208
Abstract
Elevated levels of biogenic aldehydes were reported in brains of Parkinson’s disease (PD) patients. Aldehydes are primarily detoxified by aldehyde dehydrogenases (ALDH). In PD patients, reduced ALDH1 expression and impaired complex I activity that limit the availability of the NAD+ co-factor may compromise aldehyde detoxification and play a role in PD pathogenesis. Previously, our lab showed that mice null for two Aldh isoforms known to be expressed in midbrain dopamine neurons, Aldh1a1 and Aldh2, exhibited age-related deficits in motor performance. The deficits were rescued by L-DOPA administration. Significant aldehyde accumulation in nigrostriatal pathway of Aldh1a1-/- × Aldh2-/- mice was observed, including accumulation of the lipid peroxidation product, 4-HNE, and DOPAL, the aldehyde product of dopamine metabolism. Some studies indicated that aldehyde trapping agents may be cytoprotective in conditions of increased “aldehyde load”. Therefore, we hypothesized that aldehyde trapping agents may be beneficial in PD. To test the hypothesis, we first used a PC12 cell culture model, and found that hydralazine prevented 4-HNE-induced cell death. Hydralazine (250 mg/L) was then delivered chronically in drinking water to Aldh1a1-/- × Aldh2-/- mice and improved motor performance. Reduced striatal DOPAL and midbrain 4-HNE level in Aldh1a1-/- × Aldh2-/- mice after long-term (9 month) hydralazine treatment supported our hypothesis that aldehyde trapping agents scavenge accumulating aldehydes and may provide a novel therapeutic approach to PD. As a currently FDA-approved antihypertensive treatment, hydralazine would be readily approved as a neuroprotective agent in PD.
Keywords
Biogenic aldehydes, neurotoxicity, aldehyde dehydrogenase (ALDH1A1, ALDH2), motor deficits, neurodegeneration
Introduction
Parkinson’s disease (PD) is the 2nd most prevalent ageassociated neurodegenerative disease affecting 2 to 5% of the population 65 to 85 years of age. Diagnostic criteria of PD require the presence of two or more of the cardinal motor symptoms that are resting tremor, rigidity,bradykinesia, and asymmetric onset [1]. Pathological hallmarks of PD include age-progressive degeneration of mesencephalic dopamine neurons and, in surviving neurons, the presence of Lewy bodies, of which α-synuclein is a major constituent [2]. Motor symptoms of PD are associated with loss of dopamine in the substantia nigra and striatum [3]. Numerous studies indicated that the accumulation of oxidative insults during aging contributes to PD pathogenesis. Biogenic aldehydes are toxic due to their relatively long half-life and the fact that they can cross cell membranes, acting as second toxic messengers. The hypothesis of excess “aldehyde load” has been proposed to play a role in neurodegenerative disorders [4]. Biogenic aldehydes are generated during multiple physiological processes, for example: lipid peroxidation, monoamine metabolism, and the intermediary metabolism of carbohydrates, amino acids and phospholipids [5]. Patients with Alzheimer’s disease (AD) [6-8] and PD [9, 10] have been found with high aldehyde load. Neurochemical measurements in the brains of PD patients showed elevated levels of biogenic aldehydes, including elevated 4-HNE and increased ratio of DOPAL-to-dopamine [9, 11, 12].
Aldehyde dehydrogenases (ALDH) play a major role in detoxification of biogenic aldehydes in the brain. ALDH2 deficiency has been implicated as a risk factor in AD which, like PD, is a neurodegenerative and protein aggregation disorder [13, 14]. Moreover, reduced ALDH1A1 gene expression has been reported in the substantia nigra and other tissues in PD [15-17]. Nevertheless, impaired mitochondrial complex I activity that limit the availability of the NAD+ co-factor required for aldehyde detoxification may also play a role in PD pathogenesis [18]. We have previously shown that mice null for two Aldh isoforms known to be expressed in midbrain dopamine neurons, Aldh1a1 and Aldh2, exhibited age-related deficits in motor performance on the accelerating rotarod and gait analysis. Deficits were rescued by acute L-DOPA injection. The lipid peroxidation product, 4-hydroxynonenal (4-HNE), was elevated by 30-60% in the midbrains of Aldh1a1-/- × Aldh2-/- mice. We found significant (2 to 6-fold) increased accumulation of DOPAL, the aldehyde product of dopamine metabolism, in midbrain and striatum of Aldh1a1-/- × Aldh2-/- mice. Aldehyde accumulation was associated with age-related decrease in striatal dopamine and TH+ neuronal loss in the SNpc [19].
Since increased biogenic aldehydes, DOPAL and 4-HNE, were shown to be cytotoxic in experimental models and PD patients, we postulate that enhancing the removal of aldehydes can be cytoprotective. We selected an aldehydetrapping agent, hydralazine, to test the therapeutic potential of aldehyde scavenging. Evidence has shown that hydralazine protected against aldehyde-induced cell death [20-22]. Here, we studied the aldehyde scavenging efficacy of hydralazine in a cell culture model as well as the Aldh1a1-/- × Aldh2-/- animal model of PD with elevated aldehydes by assessing motor function and neurochemical profiles after treatment. This is a proof-of-concept study that further verifies the hypothesis of aldehyde toxicity of PD and provides a novel therapeutic target for PD therapy. An increasing number of reports indicate that aldehyde trapping agents may be cytoprotective in conditions of increased “aldehyde load”. Therefore, we hypothesized that aldehyde trapping agents may be beneficial in PD. To test the hypothesis, PC12 cells were first treated with 4-HNE. 4-HNE-induced cell death was attenuated with hydralazine treatment. We then delivered hydralazine (250 mg/L) in drinking water to the Aldh1a1-/- × Aldh2-/- mice. Three or 9-month delivery of hydralazine (~ 25 mg/kg/day) produced beneficial effects on motor performance in Aldh1a1-/- × Aldh2-/- mice. The observed reduction in midbrain DOPAL levels in Aldh1a1-/- × Aldh2-/- mice following a 9-month hydralazine treatment supports our hypothesis that aldehyde-trapping agents effectively scavenge accumulating aldehydes. These findings suggest a potential novel therapeutic strategy for Parkinson’s disease (PD). As a currently FDA-approved antihypertensive treatment, hydralazine may be more readily approved as a neuroprotective agent in PD.
Materials and methods
Cell viability
The PC12 cells obtained from John Wagner [23] were grown in medium containing Dulbecco’s modified Eagle’s medium:Ham’s F12 (50:50 mix) supplemented with 5% horse serum and 10% calf serum, 2 mM L-Glutamine, 20 µg/mL penicillin and 20 µg/mL streptomycin (SAFC Biosciences, Inc., Lenexa, KS) in an incubator at 37°C under 5% CO2. The cells were seeded in 24-well multiplate at a density 1 × 105 cells/well, at least 24 hours before experimental treatments. The cells were treated with 40 µM of 4-HNE (CalBioChem Inc.) for 5 hours with 0.2% ethanol as control or with various doses of Hydralazine hydrochloride (Sigma) at 25, 50, 75, 150 and 300 µM. After treatment, cell viability was determined using the neutral red assay [24]. Briefly, cells were placed in 50 mg/mL neutral red medium (Sigma) and incubated for 2 hours. At the end of the incubation, 150 µL of elution medium (50% ethanol + 1% acetic acid) was added to each well and shaking on swing shaker for 20 min. The elution medium was transferred to a 94 well plate, and the absorbance was measured using a microplate reader at 540 nm (Multiskan MCC, Thermo Scientific, Milford, MA).
Animals and ethics statement
Animals were maintained, and experiments were conducted, in strict accordance with the Institutional Animal Care and Use Committee (IACUC), The University of Texas Health Science Center at San Antonio and the South Texas Veterans Health Care System (San Antonio, TX), and with the 1996 Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources on Life Sciences, National Research Council, National Academy of Sciences). All protocols were designed to minimize animal discomfort. Animals were group housed with five per cage in ventilated cages that were maintained at an ambient temperature of 23-25 °C and on a 12-hour light-dark cycle. Rodent bedding consisted of Sani-Chips (Harlan Teklad, Madison, WI). Mice had free access to food (Teklad 7912, Rodent sterilized diet, Harlan Teklad, Madison, WI) and water. The Aldh1a1-/- × Aldh2-/- mice used in this study were generated by crossing mice with a targeted mutation in Aldh1a1 with a line of mice carrying a mutation in Aldh2. Mice null for Aldh2 were generated by gene trap mutagenesis as we have previously reported [25] and were backcrossed to C57BL/6J mice for 10 generations. The Aldh1a1 mutant mice were generated by Gregg Duester and colleagues using a targeted deletion at exon 11 of the Aldh1a1 allele [26] and were backcrossed in our lab for 8 generations to C57BL/6J. Aldh1a1-/- mice were crossed with Aldh2-/- mice to produce mice heterozygous for both genes, Aldh1a1+/- × Aldh2+/-. Crossbreeding of Aldh1a1+/- × Aldh2+/- generated the Aldh1a1-/- × Aldh2-/- line and the wild type Aldh1a1+/+ × Aldh2+/+ line. The two lines were maintained by breeding male and female Aldh1a1-/- × Aldh2-/- or Aldh1a1+/+ × Aldh2+/+ mice. Male mice of different age groups (young, 5-8 months and middle-aged, 12-15 months), were used for the study. Body weights were monitored prior to and during behavioral testing. Treatments and behavioral tests were blinded, randomly assigned, and performed during the light cycle from 8am to 6pm if not otherwise indicated.
Hydralazine treatment
Hydralazine was delivered orally mimicking the clinical drug delivery route to evaluate its aldehyde scavenging effect. A hydralazine (Sigma) solution of 250 mg/L was prepared in drinking water. The dose was selected based on the effective dose for lowering blood pressure published previously [27-29]. The solution was prepared freshly prior to use and changed every two days in opaque water bottles to minimize light-sensitive degradation. Water intake was recorded when changing hydralazine solution and water.
Grip strength
Mice were allowed to grip the metal grids of a grip meter (Ametek Chatillon) with their forepaws, and they were gently pulled backwards by the tail until they could no longer hold the grids. The peak grip strength observed in 5 trials was recorded. Body weight and grip strength data were collected throughout the study and are presented in Figure S1.
Accelerating rotarod performance
A rotarod consists of horizontally oriented, rotating cylinder (rod) suspended above a floor that is not high enough to injure the animal, but high enough to induce avoidance of fall. Mice naturally try to stay on the rotating cylinder, or rotarod, and avoid falling to the ground. The length of time that a given animal stays on this rotating rod is a measure of their balance, coordination, physical condition, and motor-planning. The rotarod (RotaMex-5, Columbus Instruments) consists of a plastic rod, 3 cm in diameter (for mouse), with a non-slippery surface constructed from grey PVC with a knurled finish 44.5 cm above the base. This rod is divided into four equal sections (approximately 9.5 cm in width) by rounded discs, which enables four mice to walk on the rod at the same time. To test for drug effects longitudinally, mice were trained on the rotarod for 5 days until they reached steady state performance to rule out learning effect. A baseline measurement was obtained after the training. Three trials were performed on each day. During each trial, the rotarod began at an initial speed of 4 rpm and then increased gradually to 40 rpm within 300 seconds. The average latency to fall from the accelerating rod was calculated from the three trials performed on the testing day.
Automated gait analysis
Animals were tested on the ExerGait (Columbus Instruments) treadmill at a speed of 15 cm/sec for 10 sec. A digital camera recorded the reflected images of the footpads. The digital camera was operated at 80 frames per second. Automatedly collected data files were directly saved to a linked computer. Video recordings were analyzed by TreadScanTM 1.0 software (Clever Sys Inc.). The average of each parameter was collected from the strides within a 20–100 mm range.
Challenging beam traversal
Motor performance was measured with a novel beam test adapted from Fleming et al [30]. The beam was constructed from Plexiglas (Plastics Zone, Woodland Hills, CA) and consisted of four sections (25 cm each, 1 m total length), each section having a different width. The beam started at a width of 3.5 cm and gradually narrowed to 0.5 × 1 cm increments. Under-hanging ledges (1 cm width) were placed 1 cm below the upper surface of the beam. Animals were trained to traverse the length of the beam starting at the widest section and ending at the narrowest, most difficult section. The narrow end of the beam led directly into the animal's home cage. Animals received 2 days of training prior to testing, and all training was performed without the mesh grid. On the first day, animals received two assisted trials, which involved placing the animal on the beam and positioning the home cage near the animal. After two assisted trials, animals were able to traverse the entire length of the beam unassisted. Day one of training ended after all animals completed five runs across the entire length of the beam. On day 2 of training, animals were required to run five trials. To increase difficulty, on the day of the test, a mesh grid (1 cm squares) of corresponding width was placed over the beam surface leaving a ~ 1 cm space between the grid and the beam surface. The under-hanging ledges provided support for the animal to use when a limb slipped on the grid and permits assessment of a deficit chronically so that the mice do not need to use compensatory motor strategies to complete the task. Animals were then videotaped while traversing the grid-surfaced beam for a total of three trials.
Videotapes were viewed and rated in slow motion for errors, number of steps made by each animal, and time to traverse across five trials by an investigator blind to the mouse genotype. A step was counted for each gait cycle that is a forward movement of four paws. An error was counted when, during a forward movement, a limb (forelimb or hindlimb) slipped through the grid and was visible between the grid and the beam surface. An individual animal could make a maximum of four slips per step. Slips were not counted if the animal was not making a forward movement or when the animal's head was oriented to the left or right of the beam. Error per step scores, time to traverse, and number of steps were calculated for wild-type and double KO mice across all three trials and averaged.
Preparation of brain tissue
Brains were rapidly collected by decapitation following brief CO2 inhalation. For neurochemical assays, brain tissue was rapidly dissected on an ice-cold platform; tissue was then snap-frozen on dry ice and transferred to a -80 °C freezer for storage. Samples were homogenized in NP40 lysis buffer or ice-cold 0.1 M perchloric acid (HClO4) for Western or HPLC analysis, respectively.
Measurement of the aldehyde intermediate of dopamine metabolism, DOPAL
DOPAL was measured at the Catecholamine Resource Unit of the Clinical Neurocardiology Section of the NINDS according to a previously described method [12]. Samples were homogenized in an acid solution containing 20:80 parts of 0.2 M acetic acid and phosphoric acid solution. After homogenization, samples were centrifuged, and the supernatant was frozen and stored at -80°C freezer for storage until assayed for DOPAL by HPLC with electrochemical detection following batch alumina extraction. Briefly, mobile phase containing octanesulfonic acid as an ion pairing agent was pumped isocratically through a reverse phase liquid chromatographic column. Catechols were quantified by the current produced upon exposure of the eluate to flow-through electrodes set to oxidizing and then reducing potentials in series, with recording from the last electrode reflecting reversibly oxidized species.
Statistical analysis
ignificance of the effects of hydralazine treatment in PC12 cells was determined by two-way ANOVA with treatment and dosage as factors. Significance of the effects of hydralazine treatment in motor performance of Aldh-deficient mice was determined by two-way ANOVA repeated measures that assessed the effect over treatment duration with treatment and genotype as factors. Significance of the effects of hydralazine treatment in motor performance of Aldh-deficient mice was determined by two-way ANOVA with treatment and genotype as factors. Differences between individual means were assessed by the student-Newman-Keuls post-hoc comparison to assess differences between individual means.
Results
Effect of hydralazine treatment on 4-HNE-induced cell death
Figure 1 shows the effect of hydralazine on 4-HNEinduced cell death in PC12 cells. We used the neutral red uptake assay to measure 4-HNE (40 µM)-induced cell death to be 80.4% (P < 0.0001). At doses of 25, 50, 75, 150 and 300 µM hydralazine pre-treatment, a 1.6-, 2-, 3-, 3.5- and 4-fold increase in cell survival was observed, respectively.
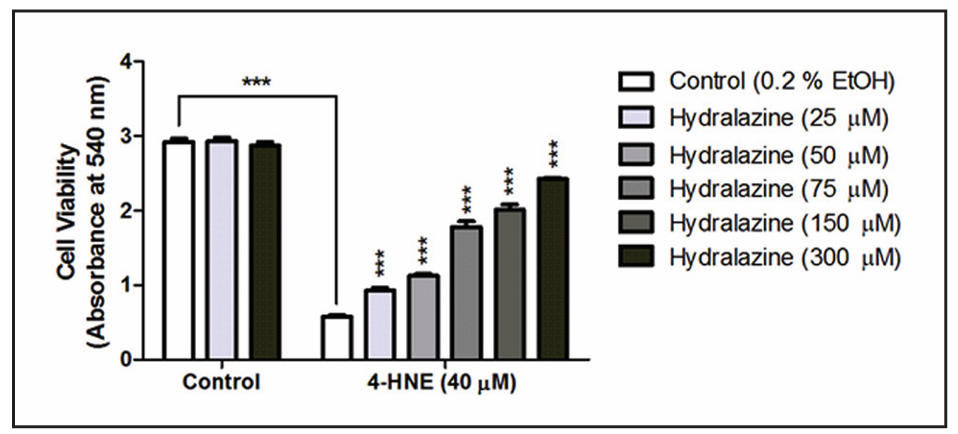
Figure 1. Effect of Hydralazine on 4-HNE induced cell death by neutral red assay. Pre-treatment of various doses of hydralazine (25, 50, 75, 150, and 300 µM) rescued 4-HNE (40 µM) induced cell death in PC12 cell culture. Data expressed as mean ± SEM; n = 3 per group. * Asterisks above bars indicate significant difference between 4-HNEtreated cells. * Asterisks above lines indicate significant difference between indicated means. ***P < 0.001.
Effect of hydralazine treatment on motor coordination
Figure 2 shows the effect of hydralazine in Aldh1a1-/- × Aldh2-/- mice on rotarod performance. Mice were treated with hydralazine at 8 and 12 months. A significant effect of genotype was observed (P < 0.001) at both age groups.
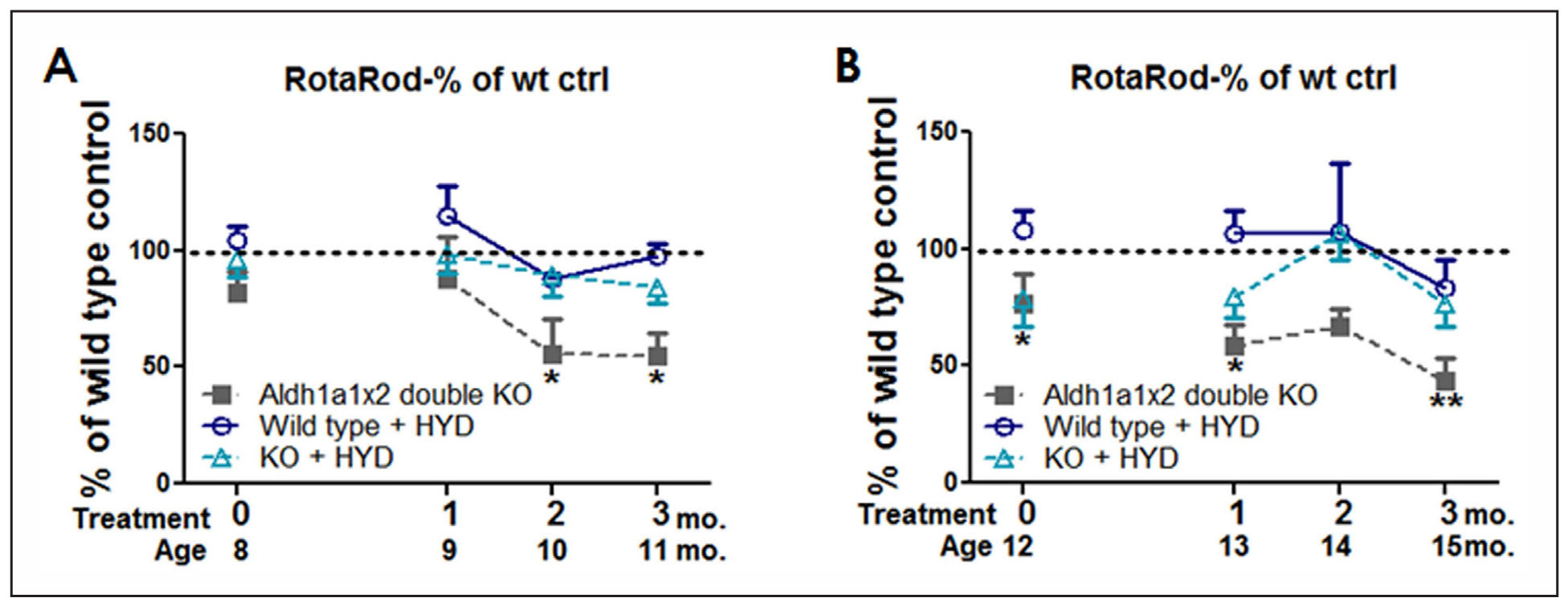
Figure 2. Hydralazine treatment for 3 months improved rotarod performance in Aldh1a1-/- ×Aldh2-/- mice. Animals were treated with 250 mg/L of hydralazine in drinking water for 3 months starting at (A) 8 months of age and (B) 12 months of age. Rotarod performance was measured longitudinally every month during the treatment. Data expressed as mean ± SEM as percentage to wild type control group. Data analyzed by two-way ANOVA repeated measures followed by SNK post-hoc comparison. *P < 0.05, **P < 0.01 compared to wild type control unless otherwise indicated; n = 4-9 per group.
Mice that started on hydralazine treatment at 12 months old had already developed impaired rotarod performance prior to treatment (Figure 2B). Significant effect of treatment (P < 0.001) was also observed in both age groups. The decreased latency to fall on the accelerating rotarod observed in the Aldh1a1-/- × Aldh2-/- group was rescued after 2 months of hydralazine treatment (wt-control vs. ko-control, P = 0.048; ko-control vs. ko-hydralazine, P = 0.040) and maintained until after 3 months of treatment. Those treated with hydralazine had significantly improved rotarod performance in comparison to those only on drinking water (P = 0.028). Mice started hydralazine treatment at 8 months of age (Figure 2A) also showed beneficial effect of hydralazine. At 10 and 11 months of age, that are after 2 and 3 months of hydralazine oral delivery, Aldh1a1-/- × Aldh2-/- mice without hydralazine treatment exhibited motor impairment that has been found earlier, while the hydralazine-treated group of Aldh1a1-/- × Aldh2-/- mice still maintained rotarod performance.
At 10 and 11 months old, Aldh1a1-/- × Aldh2-/- mice exhibit significant motor impairment on the rotarod. At the same age, Aldh1a1-/- × Aldh2-/- mice treated with hydralazine for 2 and 3 months maintained rotarod performance.
Effect of hydralazine treatment on stride length
Figure 3 shows the effect of hydralazine in Aldh1a1-/- × Aldh2-/- mice on stride length. Every two weeks, mice were placed on the treadmill ExerGait apparatus for gait analysis. Stride length, the parameter previously found to be decreased in the Aldh1a1-/- × Aldh2-/- mice, was used to evaluate the effect of hydralazine on motor function. Mice that started on hydralazine treatment at 12 months of age showed significant increase in stride length after 3 months of treatment (Figure 3B). Mice started hydralazine treatment at 8 months of age (Figure 3A) started to show decreased stride length at 9 months of age, and hydralazine oral delivery was able to maintain stride length in Aldh1a1-/- × Aldh2-/- mice with a significant difference observed after 12 weeks of treatment (wt-control vs. kocontrol, P < 0.001; ko-control vs. ko-hydralazine, P = 0.010).
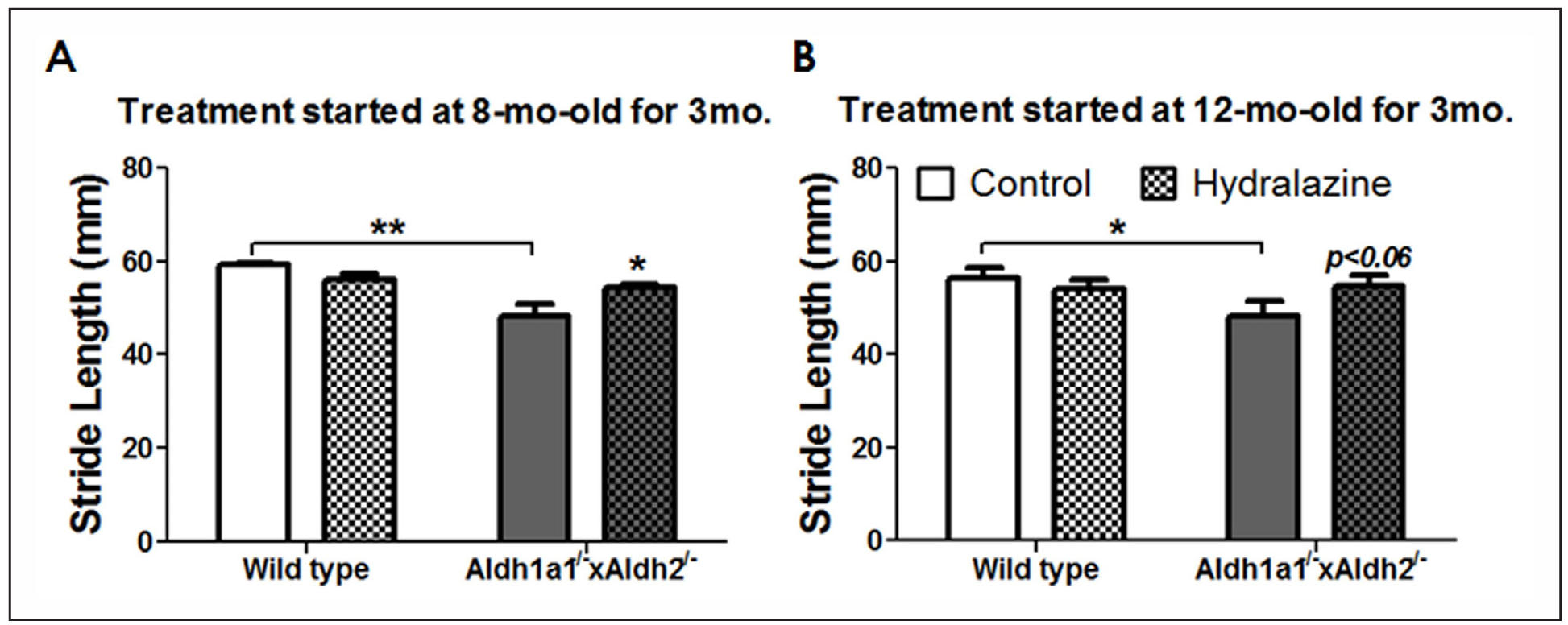
Figure 3. Hydralazine treatment for 3 months improved performance in beam test in Aldh1a1-/- × Aldh2-/- mice. Animals were treated with 250 mg/L of hydralazine in drinking water. Performance in challenging beam traversal was measured before hydralazine delivery at (A) 8 months of age and (B) 12 months of age. The effect of hydralazine on beam test performance was measured after 3 months of hydralazine treatment starting at (C) 8 months of age and (D) 12 months of age. Data expressed as mean ± SEM. Data analyzed by two-way ANOVA repeated measures followed by SNK post-hoc comparison. *P < 0.05, **P < 0.01; n = 4-9 per group.
Effect of hydralazine treatment on challenging beam traversal
Figure 4 shows the effect of hydralazine in Aldh1a1-/- × Aldh2-/- mice on the challenging beam traversal test. A significant effect of age (F1,51 = 4.346, P = 0.042) and geno type (F1,51 = 10.142, P = 0.003) was found in the baseline percent error per step measurement prior to hydralazine delivery in Aldh1a1-/- × Aldh2-/- mice. Aldh1a1-/- × Aldh2-/- animals exhibited higher rate of error per steps starting at 8 months of age (31%, P < 0.047) and 12 months of age (35.9%, P < 0.017). No changes were found in total steps and time to traverse the beam (data not shown). After 3 months of hydralazine treatment, a significant effect of treatment was found in both age groups starting hydralazine treatment at 8 (F1,21 = 9.634, P = 0.005) and 12 months old (F1,21 = 6.064, P = 0.023).
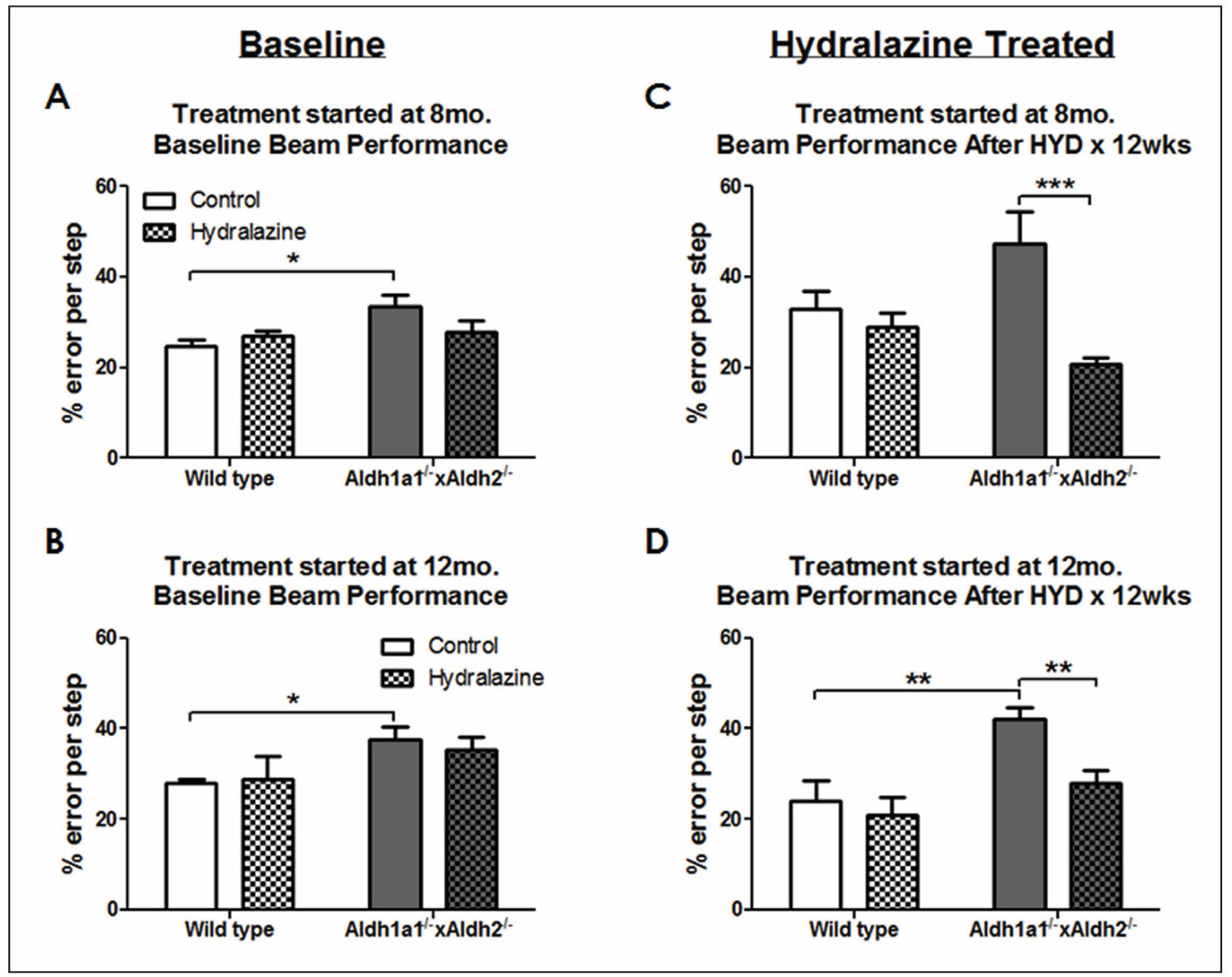
Figure 4. Cell proliferation in HGPS patients-derived fibroblasts AG11513 (A) and HGADFN271(B) untreated and treated with celastrol or AZD1981. Both fibroblast strains were treated with 0.1 µM celastrol, 10 µM AZD1981, or DMSO alone (untreated control), which was supplemented freshly every 3 or 4 days. Cell proliferation was monitored by periodical cell counting for 72 days and the cumulative population doubling levels (PDLs) were calculated. Note the differences in cumulative PDLs between the two fibroblast strains.
Effect of hydralazine treatment on aldehyde content
Figure 5 shows striatal DOPAL content of Aldh1a1-/- × Aldh2-/- mice and wild type controls with or without hydralazine oral administration. Accumulation of DOPAL was pronounced in Aldh1a1-/- × Aldh2-/- mice in the two age groups (F1,19 = 63.05, P < 0.0001; F1,22 = 61.66, P < 0.0001). However, no significant effect of treatment was found after hydralazine treatment for 3 months in 8 months old (F1,19 = 0.869, P = 0.363) or 12-month-old (F1,22 = 0.854, P = 0.366) mice.
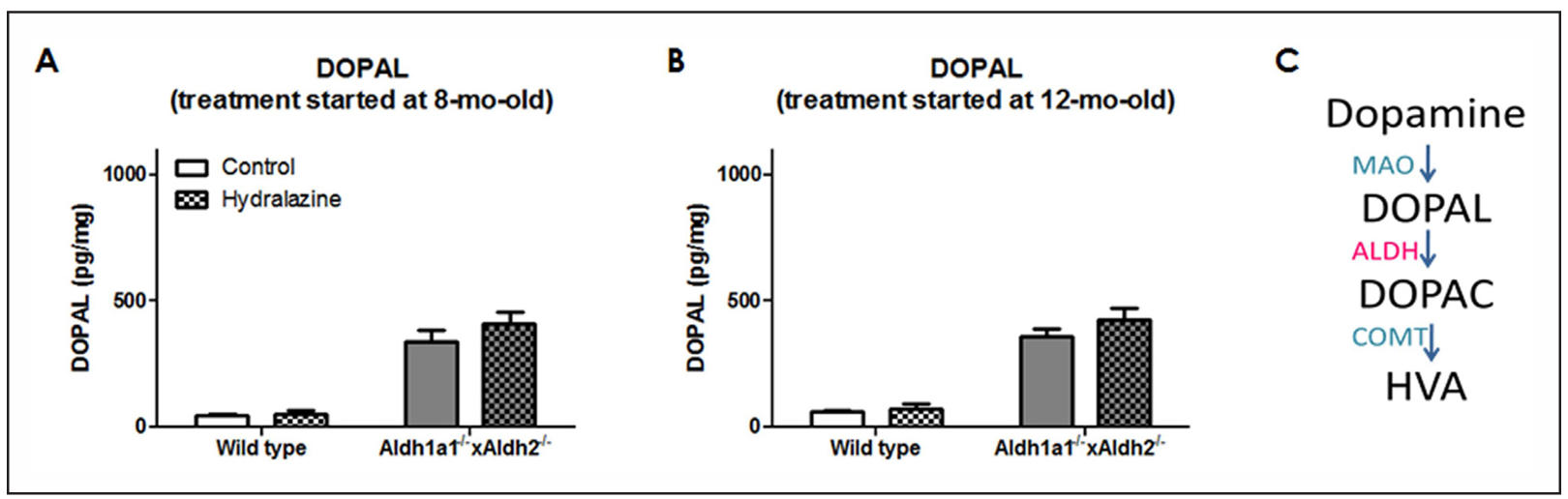
Figure 5. Effect of 3-month hydralazine treatment on striatal DOPAL content. Animals were treated with 250 mg/L of hydralazine in drinking water for 3 months starting at (A) 8 mo old and (B) 12 mo old. Striata samples were collected for HPLC analysis. Data expressed as mean ± SEM, analyzed by two-way ANOVA followed by SNK post-hoc comparison. *P < 0.05; n = 4-9 per group.
Effect of long-term hydralazine treatment on motor performance
Figure 6 shows the effect of hydralazine in Aldh1a1-/- × Aldh2-/- mice on rotarod performance (Figure 6A) and footprint gait analysis (Figure 6B). Animals were given hydralazine (250 mg/L) in drinking water for 9 months. Accelerating rotarod was tested weekly after 5-day training to evaluate motor coordination before and during hydralazine treatment. To minimize the innate fluctuation nature of behavioral tests, data are expressed as mean ± SEM as percentage to wild type control group. Rotarod data shows monthly testing result (Figure 6A).
Figure 6B shows the effect of 9-month hydralazine delivery in Aldh1a1-/- × Aldh2-/- mice on stride length. Footprints were measured to obtain stride length. A significant effect of genotype was found in Aldh1a1-/- × Aldh2-/- mice without hydralazine treatment compared to non-treated wild type controls (P = 0.025).
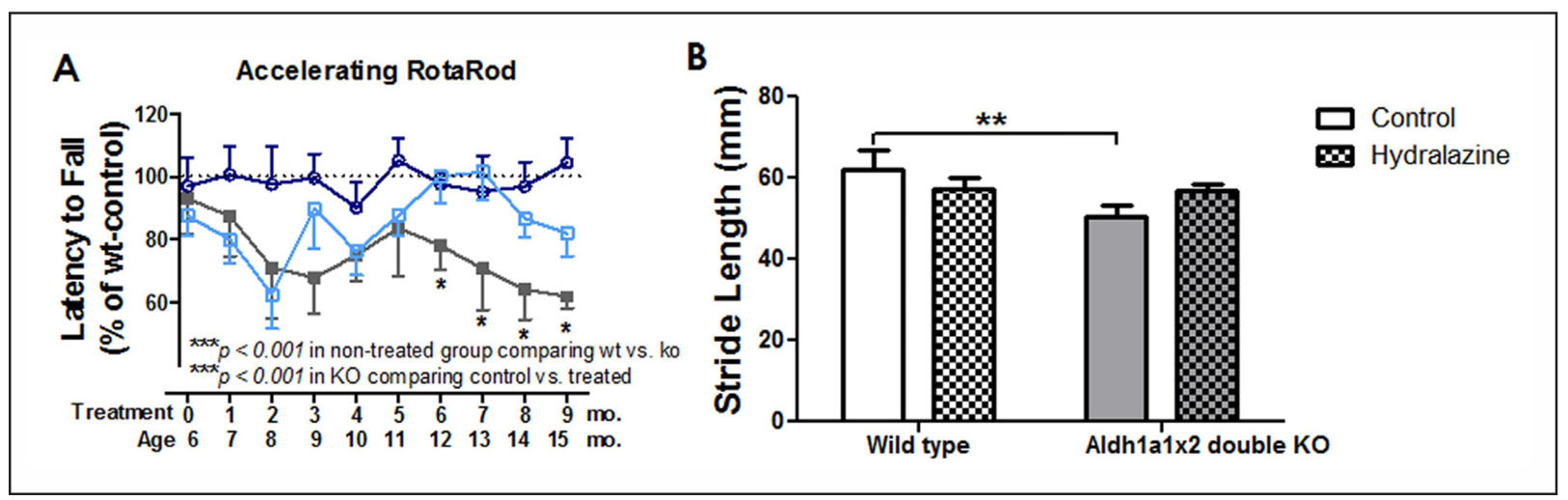
Figure 6. Hydralazine treatment for 9 months improved motor performance in Aldh1a1-/- × Aldh2-/- mice. Animals were treated with 250 mg/L of hydralazine in drinking water. (A) Accelerating rotarod and (B) gait analyses were measured after 9 months of hydralazine treatment. Data expressed as (A) mean ± SEM as percentage to wild type control group and (B) mean ± SEM. Aldh1a1x2 double KO mice are plotted in solid grey squares, wild type + HYD in blue open circles, and KO + HYD in light blue open squares. Data analyzed by two-way ANOVA repeated measures followed by SNK post-hoc comparison. *P < 0.05, **P < 0.01; n = 4-9 per group.
Effect of long-term hydralazine treatment on aldehyde content
Figure 7 shows the effect of hydralazine treatment for 9 months on striatal DOPAL level. Animals were sacrificed at 15 months of age after 9 months of hydralazine treatment. A significant effect of genotype on striatal DOPAL accumulation was found in Aldh1a1-/- × Aldh2-/- mice without hydralazine treatment compared to non-treated wild type controls (F1,21 = 83.053, P < 0.001). A significant effect of hydralazine treatment (F1,21 = 4.086, P = 0.05) was found in Aldh1a1-/- × Aldh2-/- mice (P = 0.032) but not in the wild type mice (P = 0.607).
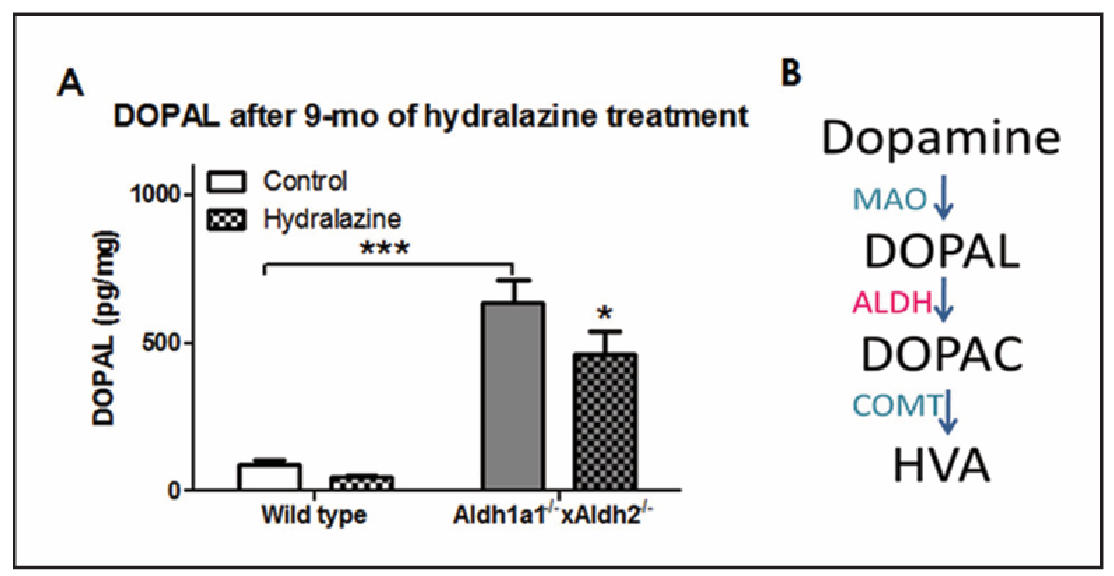
Figure 7. Hydralazine treatment for 9 months decreased striatal DOPAL content. Animals were treated with 250 mg/L of hydralazine in drinking water for 9 months starting at 6 months old. Striata samples were collected for HPLC analysis. Data expressed as mean ± SEM, analyzed by two-way ANOVA followed by SNK post-hoc comparison. *P < 0.05; n = 4-9 per group.
Discussion
This study describes the effect of hydralazine on motor behavior and aldehyde scavenging in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases. We previously reported that the Aldh1a1-/- × Aldh2-/- mouse model mimicked PD-like motor deficits and exhibited neurochemical manifestations accompanied by elevated levels of biogenic aldehydes (i.e., DOPAL and 4-HNE) in the nigrostriatum [19]. In vitro, DOPAL and 4-HNE, has been reported to induce cell death [31, 32]. Hydralazine was approved by the FDA as an antihypertensive agent in 1953 [33]. It decreases blood pressure by direct vasodilation. Moreover, hydralazine condensation with various toxic biogenic aldehydes to form a nontoxic hydrazone is noteworthy [34]. In the current study, treatment with hydralazine at various doses prevented 4-HNE-induced cell death in a dose dependent manner. Although the 4-HNE-induced cell death was still significant after 300 µM of hydralazine treatment compared to non-treated controls, there was a 4.21-fold increase in cell survival compared to 4-HNE treated PC12 cells. This is consistent with the result that hydralazine rescued PC12 cells from acrolein-induced cell death. Liu-Snyder and coworkers [2] showed in various assays that hydralazine not only rescued acrolein-induced cell death, but also restored depleted intracellular ATP and glutathione. In addition, several ex vivo studies reported that hydralazine mitigated acrolein-mediated damage [35, 36].
To investigate the protective effect of hydralazine In vitro, we treated Aldh1a1-/- × Aldh2-/- mice with 250 mg/L of hydralazine in drinking water and assessed hydralazine efficacy using behavioral tests sensitive to changes in dopaminergic activity. Long term treatment of hydralazine in drinking water improved motor performance measured by accelerating rotarod, gait analysis and challenging beam test. Aldh1a1-/- × Aldh2-/- animals treated for 9 months showed prolonged beneficial effect on motor coordination. Treatment with hydralazine for 3 months, initiated at 8 and 12 months of age, improved the deleterious effects observed on motor function but had no effect on DOPAL content. Long-term hydralazine treatment has shown sustained neuroprotective effects, particularly in our model mouse model of Parkinson’s disease. So far, there have not been many functional studies of hydralazine on neurodegenerative animal models. One report showed that treatment with hydralazine at 1 mg/kg for 30 days delayed the onset and reduced motor impairment in experimental autoimmune enchephalomyelitis mice, a model for multiple sclerosis [37]. Our finding that hydralazine improved PDlike motor deficit is novel. This is consistent with previous studies that shows that hydralazine renders protection against acroleinand allyl alcohol-induced hepatotoxicity as a strong aldehyde scavenging agent [21]. Hydralazine or hydralazine-loaded nanoparticles also protected against acrolein-mediated cell injury [22, 38].
Conclusions
In summary, we determined the effect of aldehyde scavenging agent, hydralazine, in Aldh1a1 and Aldh2 double knockout mice. In vitro and in vivo administration of hydralazine not only ameliorated aldehyde-induced toxicity, but also exerted beneficial outcomes in motor functions. In future directions we will (1) study hydralazine pharmacokinetics in the central nervous system and (2) explore other potential aldehyde scavenging compounds in addition to hydralazine. Because of the wide variety of pathological processes associated with oxidative stress and subsequent aldehyde accumulation, research into aldehyde trapping agents may provide an alternative approach that can benefit degenerative diseases. In addition, the repurposing of an FDA-approved drug may fast track the use of hydralazine in PD therapy.
Declarations
Acknowledgments
We thank Shou-Shu Wang, Xiang (Vicky) Bai, and Vanessa Martinez for their effort and contribution in running behavioral experiments. We thank Vivian Diaz and the Aging Animal Models and Longevity Assessment Core (a part of the San Antonio Nathan Shock Center) for breeding and maintaining the mouse colony.
Authorship Contributions
Participated in research design: Wey, Fernandez and Strong. Conducted experiments: Wey, Martinez, Wang and Sullivan. Contributed new reagents or analytic tools: Sullivan and Goldstein. Performed data analysis: Wey and Sullivan. Wrote or contributed to the writing of the manuscript: Wey, Martinez, Goldstein, Fernandez and Strong.
Financial support and sponsorship
This work was supported by grants from the Veteran Affairs Office of Research & Development Senior research career scientist award Grant12709081(to R.S/E.F.) and the National Institute of Aging pre-doctoral training grant T32 AG021890- 08 (to M.C.W.).
References
1. Gelb D, Oliver E, & Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol, 1999, 56(1): 33-39. [Crossref]
2. Spillantini M, Schmidt M, Lee V, Trojanowski J, Jakes R, & Goedert M. Alpha-synuclein in Lewy bodies. Nature, 1997, 388(6645): 839-840. [Crossref]
3. Carlsson A, Lindqvist M, & Magnusson T. 3,4-Dihydroxyphenylalanine and 5-Hydroxytryptophan as reserpine antagonists. Nature, 1957, 180(4596): 1200-1200. [Crossref]
4. Wood P. Neurodegeneration and aldehyde load: from concept to therapeutics. J Psychiatry Neurosci, 2006, 31(5): 296-297.
5. Marchitti S, Deitrich R, & Vasiliou V. Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol Rev, 2007, 59(2): 125-150. [Crossref]
6. Calingasan N, Uchida K, & Gibson G. Protein-bound acrolein: a novel marker of oxidative stress in Alzheimer's disease. J Neurochem, 1999, 72(2): 751-756. [Crossref]
7. Picklo M, Montine T, Amarnath V, & Neely M. Carbonyl toxicology and Alzheimer's disease. Toxicology and Applied Pharmacology, 2002, 184(3): 187-197. [Crossref]
8. Williams T, Lynn B, Markesbery W, & Lovell M. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in mild cognitive impairment and early Alzheimer's disease. Neurobiol Aging, 2006, 27(8): 1094-1099. [Crossref]
9. Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman E, & Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci USA, 1996, 93(7): 2696-2701. [Crossref]
10. Jenner P. Oxidative stress in Parkinson's disease. Ann Neurol, 2003, 53 Suppl 3: S26-36; discussion S36-28. [Crossref]
11. Mattammal M, Chung H, Strong R, & Hsu F. Confirmation of a dopamine metabolite in parkinsonian brain tissue by gas chromatography-mass spectrometry. J Chromatogr, 1993, 614(2): 205-212. [Crossref]
12. Goldstein D, Sullivan P, Holmes C, Kopin I, Basile M, & Mash D. Catechols in post-mortem brain of patients with Parkinson disease. Eur J Neurol, 2011, 18(5): 703-710. [Crossref]
13. Wang B, Wang J, Zhou S, Tan S, He X, Yang Z, et al. The association of mitochondrial aldehyde dehydrogenase gene (ALDH2) polymorphism with susceptibility to lateons et alzheimer's disease in Chinese. J Neurol Sci, 2008, 268(1-2): 172-175. [Crossref]
14. Michel T, Gsell W, Käsbauer L, Tatschner T, Sheldrick A, Neuner I, et al. Increased activity of mitochondrial aldehyde dehydrogenase (ALDH) in the putamen of individuals with Alzheimer's disease: a human postmortem study. J Alzheimers Dis, 2010, 19(4): 1295-1301. [Crossref]
15. Galter D, Buervenich S, Carmine A, Anvret M, & Olson L. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson's disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis, 2003, 14(3): 637-647. [Crossref]
16. Mandel S, Grunblatt E, Riederer P, Amariglio N, JacobHirsch J, Rechavi G, et al. Gene expression profiling of sporadic Parkinson's disease substantia nigra pars compacta reveals impairment of ubiquitin-proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann NY Acad Sci, 2005, 1053: 356-375. [Crossref]
17. Grünblatt E, Mandel S, Jacob-Hirsch J, Zeligson S, Amariglo N, Rechavi G, et al. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J Neural Transm (Vienna), 2004, 111(12): 1543-1573. [Crossref]
18. Meyer M, Mosely D, Amarnath V, & Picklo M, Sr. Metabolism of 4-hydroxy-trans-2-nonenal by central nervous system mitochondria is dependent on age and NAD+ availability. Chem Res Toxicol, 2004, 17(9): 1272-1279. [Crossref]
19. Wey M, Fernandez E, Martinez P, Sullivan P, Goldstein D, & Strong R. Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: implications for Parkinson's disease. PLoS One, 2012, 7(2): e31522. [Crossref]
20. Burcham P, Fontaine F, Kaminskas L, Petersen D, & Pyke S. Protein adduct-trapping by hydrazinophthalazine drugs: mechanisms of cytoprotection against acrolein-mediated toxicity. Mol Pharmacol, 2004, 65(3): 655-664. [Crossref]
21. Burcham P, Kaminskas L, Fontaine F, Petersen D, & Pyke S. Aldehyde-sequestering drugs: tools for studying protein damage by lipid peroxidation products. Toxicology, 2002, 181-182: 229-236. [Crossref]
22. Liu-Snyder P, Borgens R, & Shi R. Hydralazine rescues PC12 cells from acrolein-mediated death. J Neurosci Res, 2006, 84(1): 219-227. [Crossref]
23. Van Buskirk R, Corcoran T, & Wagner J. Clonal variants of PC12 pheochromocytoma cells with defects in cAMP-dependent protein kinases induce ornithine decarboxylase in response to nerve growth factor but not to adenosine agonists. Mol Cell Biol, 1985, 5(8): 1984-1992. [Crossref]
24. Fautz R, Husein B, & Hechenberger C. Application of the neutral red assay (NR assay) to monolayer cultures of primary hepatocytes: rapid colorimetric viability determination for the unscheduled DNA synthesis test (UDS). Mutat Res, 1991, 253(2): 173-179. [Crossref]
25. Chen Z, Foster M, Zhang J, Mao L, Rockman H, Kawamoto T, et al. An essential role for mitochondrial aldehyde dehydrogenase in nitroglycerin bioactivation. Proc Natl Acad Sci USA, 2005, 102(34): 12159-12164. [Crossref]
26. Fan X, Molotkov A, Manabe S, Donmoyer C, Deltour L, Foglio M, et al. Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol Cell Biol, 2003, 23(13): 4637-4648. [Crossref]
27. Qi G, Jia L, Li Y, Bian Y, Cheng J, Li H, et al. Angiotensin II infusion-induced inflammation, monocytic fibroblast precursor infiltration, and cardiac fibrosis are pressure dependent. Cardiovasc Toxicol, 2011, 11(2): 157-167. [Crossref]
28. Cassis L, Gupte M, Thayer S, Zhang X, Charnigo R, Howatt D, et al. ANG II infusion promotes abdominal aortic aneurysms independent of increased blood pressure in hypercholesterolemic mice. Am J Physiol Heart Circ Physiol, 2009, 296(5): H1660-1665. [Crossref]
29. Marvar P, Thabet S, Guzik T, Lob H, McCann L, Weyand C, et al. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ Res, 2010, 107(2): 263-270. [Crossref]
30. Fleming S, Salcedo J, Fernagut P, Rockenstein E, Masliah E, Levine M, et al. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J Neurosci, 2004, 24(42): 9434-9440. [Crossref]
31. Chen J, Wang L, Chen Y, Sternberg P, & Cai J. Phosphatidylinositol 3 kinase pathway and 4-hydroxy-2-nonenalinduced oxidative injury in the RPE. Invest Ophthalmol Vis Sci, 2009, 50(2): 936-942. [Crossref]
32. Mattammal M, Haring J, Chung H, Raghu G, & Strong R. An endogenous dopaminergic neurotoxin: implication for Parkinson's disease. Neurodegeneration, 1995, 4(3): 271-281. [Crossref]
33. Slim H, Black H, & Thompson P. Older blood pressure medications-do they still have a place? Am J Cardiol, 2011, 108(2): 308-316. [Crossref]
34. O'Donnell J, Proveaux W, & Ma J. High-performance liquid chromatographic studies of reaction of hydralazine with biogenic aldehydes and ketones. J Pharm Sci, 1979, 68(12): 1524-1526. [Crossref]
35. Hamann K, Durkes A, Ouyang H, Uchida K, Pond A, & Shi R. Critical role of acrolein in secondary injury following ex vivo spinal cord trauma. J Neurochem, 2008, 107(3): 712-721. [Crossref]
36. Hamann K, Nehrt G, Ouyang H, Duerstock B, & Shi R. Hydralazine inhibits compression and acrolein-mediated injuries in ex vivo spinal cord. J Neurochem, 2008, 104(3): 708-718. [Crossref]
37. Leung G, Sun W, Brookes S, Smith D, & Shi R. Potassium channel blocker, 4-aminopyridine-3-methanol, restores axonal conduction in spinal cord of an animal model of multiple sclerosis. Exp Neurol, 2011, 227(1): 232-235. [Crossref]
38. Cho Y, Shi R, & Ben Borgens R. Chitosan nanoparticlebased neuronal membrane sealing and neuroprotection following acrolein-induced cell injury. J Biol Eng, 2010, 4(1): 2-14. [Crossref]
Supplementary
Figure S1. Hydralazine treatment did not affect body weight and muscle strength. Data are expressed as the mean ± SEM. Animals were treated with 250 mg/L of hydralazine in drinking water for 3 months or 9 months starting at middle age or young age. Body weight and grip strength were measured monthly prior to behavioral assessments.