Open Access | Research
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
Integrative bioinformatics and molecular docking unveil multi-target therapeutic mechanisms of Coptidis Rhizoma (CR) against Lewy body dementia (LBD): an in-silico approach
* Corresponding author: Pranab Dev Sharma
Mailing address: Biotechnology program, Department of Mathematics and Natural Science, BRAC University, Dhaka-1212, Bangladesh.
Email: pranab.dev.sharma@g.bracu.ac.bd
Received: 28 March 2025 / Revised: 23 April 2025 / Accepted: 07 May 2025 / Published: 30 September 2025
DOI: 10.31491/APT.2025.09.181
Abstract
Background: Lewy body dementia (LBD) is a complex neurodegenerative
disorder characterized by the presence of Lewy bodies in the brain, leading to
cognitive decline and motor symptoms. The exploration of potential therapeutic
mechanisms is crucial for developing effective treatments. Coptidis Rhizoma (CR) has
been traditionally used in herbal medicine, and its compounds may offer novel
therapeutic avenues for LBD.
Materials and Methods: In this study, an integrative bioinformatics
approach was employed to investigate the molecular interactions between CR compounds
and LBD-associated targets. The authors collected relevant targets from the OMIM and
NCBI databases and utilized bioinformatics tools for protein-protein interaction (PPI)
network analysis, gene ontology, and KEGG pathway enrichment. Molecular docking
simulations were conducted using PyRx software to evaluate binding interactions
between CR compounds and key proteins associated with LBD.
Results: The analysis revealed 20 critical hub genes, including MAPT,
GBA, SNCA, TARDBP, and PRKN, which play significant roles in neurodegeneration.
Key processes involved in LBD, such as neuronal apoptosis regulation, oxidative
stress response, and synaptic transmission, were identified. The molecular docking
results indicated compelling interactions, with Obacunone exhibiting a binding
energy of -9.4 kcal/mol for GBA and -9.2 kcal/mol for PRKN, suggesting its
strong affinity for these targets. Similarly, Coptisine demonstrated high
binding potential with GBA (-9.4 kcal/mol), while Worenine showed notable
interaction with SNCA (-7.4 kcal/mol), reinforcing their therapeutic relevance.
Conclusion: This computational study provides valuable
insights into the therapeutic mechanisms of CR compounds for LBD.
The findings underscore the importance of experimental validation
to confirm the predicted interactions and therapeutic potential,
paving the way for future in vitro and in vivo investigations to
address this challenging neurodegenerative disorder.
Keywords
Lewy body dementia (LBD), Coptidis Rhizoma (CR), neurodegenerative disorder, network pharmacology, molecular docking
Introduction
Lewy body dementia (LBD) is a complex and devastating neurodegenerative disorder
characterized by progressive cognitive decline, motor dysfunction, and significant
neurological impairments [1-3]. However, given its severe effects on patient quality
of life and sustained burden on the healthcare system, current treatment options are
limited, highlighting a desperate need for further research initiatives that can help
to elucidate the fundamental molecular drivers of the disease [4, 5].
Lewy body dementia (LBD) is defined by the abnormal deposition of alpha-synuclein protein
aggregates in neuronal cells, called Lewy bodies [6]. These pathological inclusions interfere
with essential cellular processes, resulting in neuronal impairment and gradual
neurodegeneration [7]. The complex interplay among genetic, environmental, and molecular
factors contributes to the heterogeneous etiology of the disease and hinders the development
of classical therapeutic strategies [8, 9].
Traditional Chinese Medicine (TCM) presents a promising approach for discovering new
therapeutic compounds with potential neuroprotective effects [10, 11]. Coptidis Rhizoma (CR),
an ancient medicinal herb with diverse pharmacological activities, has demonstrated
anti-inflammatory, antioxidative, and neuroprotective activities [12, 13]. C. Rhizoma
also known as Huanglian, is a traditional Chinese medicinal herb derived from the dried
rhizome of Coptis chinensis and related species [14]. However, the detailed molecular
mechanisms driving its potential therapeutic significance in LBD are not well characterized.
Network pharmacology and molecular docking are powerful computational methodologies that
have recently provided us with unprecedented insight into disease mechanisms and potential
therapeutic interventions [15, 16]. We combined an integrative bioinformatics approach to
thoroughly explore the molecular interactions of the Coptidis Rhizoma compounds and
biologically relevant targets in LBD. Using computational methods, including protein-protein
interaction networks, gene ontology analysis, pathway enrichment, and molecular docking
simulations, we aim to:
• Use signature function to identify potential therapeutic compounds from Coptidis Rhizoma
• Investigate molecular mechanisms of engagement with LBD-binding targets
• Identify mechanisms by which to treat or modify Lewy body dementia
Our study provides the systematic investigation of the complex molecular networks
underlying LBD, taking advantage of the extensive medicinal properties of traditional
herbal medicine. By connecting established knowledge with modern computational biology,
we hope to offer an original addition to putative treatment approaches for this recalcitrant
neurodegenerative disease.
Methods and materials
Target collection for Lewy body dementia (LBD)
To get the targets connected to Lewy body dementia, OMIM (Online Mendelian Inheritance in Man) database (https://www.omim.org/, accessed on 26 March 2025) and NCBI (National Center for Biotechnology Information) database (https://www.ncbi.nlm.nih.gov/, accessed on 26 March 2025) [17, 18]. “Lewy body dementia” was used as the keyword for searching. File with disease related genes (Targets) was downloaded for further process. The collected target genes then inserted in the Uniport (https://www.uniprot.org/, accessed on 26 March 2025) database to determine the targets of Homo sapiens [19].
Molecule selection of Coptidis Rhizoma (CR)
TCMSP which is Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform (https://tcmsp-e.com/tcmsp.php, accessed on 26 March 2025) was utilized to find out the active compounds present in Coptidis Rhizoma (CR) [20]. TCMSP is very useful because it captures relationship between drugs, targets, and diseases. Compounds with OB > 30% and DL > 0.18 were selected for following process. Moreover, molecular weight < 500 g/mol, H-bond acceptors < 10, H-bond donor < 5, topological polar surface area < 150, and Moriguchi octanol–water partition coefficient ≤ 4.15 were the criteria that followed in compound selection for that plant [19]. SwissADME (http://www.swissadme.ch/, accessed on 26 March 2025) tool was utilized to calculate those values [21].
Target collection for selected compounds/molecules
We applied CTD (Comparative Toxicogenomics Database) (https://ctdbase.org/, accessed on 26 March 2025), and Swiss Target Prediction tool (http://www.swisstargetprediction.ch/, accessed on 26 March 2025), for obtaining the targets for the chosen molecules [22, 23]. The name of each molecule was used to search genes in CTD and the smile of each compound was applied in Swiss Target Prediction tool to get targets. After that the collected target genes were inserted in the Uniport (https://www.uniprot.org/, accessed on 26 March 2025) database to determine the targets of Homo sapiens [24].
Identification of common targets
Venny 2.1.0 tool (https://bioinfogp.cnb.csic.es/tools/venny/, accessed on 26 March 2025) was used to identify the common targets for both disease and molecules [25]. Those intersect targets were considered as the significant relationship between drug molecules and disease.
Protein-protein interaction network
We applied the String database (https://string-db.org/, accessed on 26 March 2025) for a PPI network among those intersect targets by specifying “Homo sapiens” as the species with a high required score [26]. To visualize the interaction network, Cytoscapes 3.10.3 software was applied with cytoHubba plugin to easily recognize the targets with highest connections [27]. From that plugin, we got top twenty targets based on their degree value. Because the proteins (targets) which have high degree value are known as hub proteins and they are essential for different biological processes and key regulations in cellular pathways [28].
Gene ontology (GO) with pathway analysis
The intersection targets of the molecules from Coptidis Rhizoma (CR) and Lewy body dementia (LBD) were evaluated using the DAVID platform (https://david.ncifcrf.gov/, accessed on 26 March 2025) online for analyzing the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment investigations [29]. This tool helps to study on Molecular Functions (MFs), Cellular Components (CCs), Biological Processes (BPs), and KEGG pathways. The gene list was added with specifying Homo sapiens and the identifier was set into official gene symbol. The top 10 GO items and top 10 KEGG pathways were selected based on statistical significance (p < 0.05) and visualized using bioinformatics web platforms (http://www.bioinformatics.com.cn/, accessed on 26 March 2024).
MCODE Analysis
For grouping associated proteins and for developing functional modules using molecular complex detection (MCODE), the Metascape (https://metascape.org/, accessed on 26 March 2024) tool was utilized [30]. MCODE analysis identified protein connection complexes and classified them into gene clusters where each target has the same or comparable gene functions. Furthermore, transcription factors were predicted that control additional targets by providing information on target relationships. MCODE clustering analysis was performed for common targets based on a minimum overlap of 3, cutoff of P-value 0.05, and 1.5 for the threshold enrichment [31].
Target-pathway network construction
The 21 pathways resulting from the KEGG enrichment analysis of CR and LBD were visualized using Cytoscape 3.10.3 software to create a target–pathway network diagram. Network topology parameters were examined, with targets ranked by degree value. The top 20 targets from this analysis were intersected with those identified in the PPI network to find out common targets.
Key target identification
The recognized final targets from the intersection of PPI network and target to pathway network were used to find out key targets by comparing among different topological properties of those intersected targets. For that reason, we again used the STRING (https://string-db.org/, accessed on 26 March 2025) online platform to create a PPI network among those genes (targets) [32]. To visualize and analyze the network, Cytoscape along with Cytohubba plugin was used. We selected five topological properties properties Maximal Clique Centrality (MCC), Density of Maximum Neighborhood Component (DMNC), Bottle-Neck, Closeness, and Degree value and utilized molbiotools (https://molbiotools.com/listcompare.php, accessed on 26 March 2025) for comparing all five properties.
Molecular docking
The recognized final targets from the intersection of PPI network and target to pathway network were used to find out key targets by comparing among different topological properties of those intersected targets. For that reason, we again used the STRING (https://string-db.org/, accessed on 26 March 2025) online platform to create a PPI network among those genes (targets) [32]. To visualize and analyze the network, Cytoscape along with Cytohubba plugin was used. We selected five topological properties properties Maximal Clique Centrality (MCC), Density of Maximum Neighborhood Component (DMNC), Bottle-Neck, Closeness, and Degree value and utilized molbiotools (https://molbiotools.com/listcompare.php, accessed on 26 March 2025) for comparing all five properties.
Molecular docking
Protein and compound preparation
First of all, each protein was added into Discovery Studio software for removing
heteroatoms such as water and other ligands. After that, those proteins were inserted
into Chimera X 1.19 software for further preparation [33]. In this process, hydrogens
were added, charges and missing atoms were added, and the prepared proteins were saved
as a PDB file for docking [34]. For docking, we used PyRx software, which has the
accessibility of Autodock Vina. Each protein was added there and converted into a
macromolecule, and the next grid box for each protein was adjusted for docking [35].
The saved SDF files of compounds were directly added into PyRx for docking since
PyRx has access to Open Babel [36, 37]. The charge of each compound was minimized,
and those minimized charged compounds were converted into a PDBQT file to continue
the docking process [38].
Protein-ligand binding
The binding sites/active sites for each target were identified by using Discovery Studio’s predictions and the grid boxes optimized around the binding areas suggested by the software [39-41]. Active sites help to adjust the grid box according to protein. Discovery Studio is a useful software and helped to find out the active sites quickly [42]. While binding affinity is an important parameter, ligand interactions also depend on chemical properties, steric interactions, and protein flexibility, which influence binding to specific residues [43-45]. The grid box was set into center: x = -3.6673, y = -19.8314, z = -2.5260 and dimensions: x = 28.6315, y = 33.3054, z = 25.0000 for GBA (PDB ID: 6TA3). The grid box was set into center: x = 24.0622, y = -6.0253, z = -24.0138 and dimensions: x = 25.0259, y = 17.0974, z = 18.8013 for MAPT (PDB ID: 2MZ7). The box was selected into center: x = 3.9566, y = 1.4492, z = 2.4175 and dimensions: x = 17.3632, y = 22.0490, z = 17.3590 for TARDBP (PDB ID: 2CQG). The box was fixed into center: x = 237.5219, y = 82.2100, z = -13.4109 and dimensions: x = 32.4387, y = 26.0723, z = 39.6527 for SNCA (PDB ID: 1XQ8). Finally, for PRKN (PDB ID: 5C1Z) the grid box was fixed into center: x = -14.7309, y = 15.0906, z = 8.3024 and dimensions: x = 36.3893, y = 35.0225, z = 25.0000.
Results
Targets for disease
To identify potential disease-associated targets for Lewy body dementia, we collected 331 targets from the OMIM and NCBI databases. After filtering duplicate entries, 228 unique targets were identified and analyzed further to understand their potential relevance to disease pathology.
Molecule selection
The selection process for active molecules in Coptidis Rhizoma followed specific pharmacokinetic and drug-likeness criteria. A total of 12 molecules met the required threshold for oral bioavailability and drug-likeness, ensuring their potential effectiveness in targeting disease-related pathways (Table 1, Figure 1). Because Lipinski’s Rule of Five defines key physicochemical constraints for oral drug absorption, including molecular weight below 500 Da, fewer than 5 hydrogen bond donors, fewer than 10 hydrogen bond acceptors, and an octanol-water partition coefficient (log P) under 5 [46]. Additionally, topological polar surface area (TPSA) below 140 Ų enhances membrane permeability and blood-brain barrier penetration, which is critical for central nervous system drugs [47, 48]. Furthermore, OB > 30% signifies that the compound exhibits greater than 30% oral bioavailability, meaning effective absorption into systemic circulation via oral administration [49]. DL > 0.18 signifies drug-likeness, such that the compound displays characteristics associated with drugs, and it can be a candidate for drug development [50]. The requirements select molecules with favorable pharmacokinetics and drug-like activity to enhance drug discovery success chances [51].
Table 1.
Selected molecules on different pharmacokinetic properties.
| Molecule name | Molecular weight | H-bond donor | H-bond acceptor | TPSA | MlogP | OB% | DL |
|---|---|---|---|---|---|---|---|
| Worenine | 334.351 | 0 | 4 | 40.8 | 3.11622 | 45.83 | 0.87 |
| Coptisine | 320.324 | 0 | 4 | 40.8 | 2.8078 | 30.67 | 0.86 |
| Berlambine | 351.358 | 0 | 6 | 58.92 | 2.9705 | 36.68 | 0.82 |
| Epiberberine | 336.367 | 0 | 4 | 40.8 | 3.0963 | 43.09 | 0.78 |
| Berberine | 336.367 | 0 | 4 | 40.8 | 3.0963 | 36.86 | 0.78 |
| (R)-Canadine | 339.391 | 0 | 5 | 40.16 | 3.088 | 55.37 | 0.77 |
| Obacunone | 454.519 | 0 | 7 | 95.34 | 3.9246 | 43.29 | 0.77 |
| Berberrubine | 357.793 | 1 | 4 | 51.8 | -0.2027 | 35.74 | 0.73 |
| Palmatine | 352.41 | 0 | 4 | 40.8 | 3.3848 | 64.6 | 0.65 |
| Quercetin | 302.238 | 5 | 7 | 131.36 | 1.988 | 46.43 | 0.28 |
| Moupinamide | 313.353 | 3 | 4 | 78.79 | 2.4785 | 86.71 | 0.26 |
| Magnograndiolide | 266.337 | 2 | 4 | 66.76 | 1.4062 | 63.71 | 0.19 |
The 12 bioactive compounds from Coptidis Rhizoma (CR) were selected based on a set of pharmacokinetic and drug-likeness criteria to ensure their potential as central nervous system-active agents. Key selection parameters included a molecular weight below 500 g/moL, in accordance with Lipinski’s Rule of Five, which supports drug absorption and permeation. The number of hydrogen bond donors and acceptors was restricted to fewer than 5 and 10, respectively, to maintain favorable pharmacokinetics. The topological polar surface area (TPSA) was required to be under 150 Ų, a crucial factor for predicting the compounds' ability to cross the blood-brain barrier. Lipophilicity was assessed using the Moriguchi octanol-water partition coefficient (MlogP), with an optimal threshold of ≤ 4.15. Additionally, compounds with oral bioavailability (OB) greater than 30% were prioritized to ensure effective systemic absorption. Lastly, a drug-likeness (DL) score above 0.18 was used to confirm structural and physicochemical features consistent with known drug compounds.
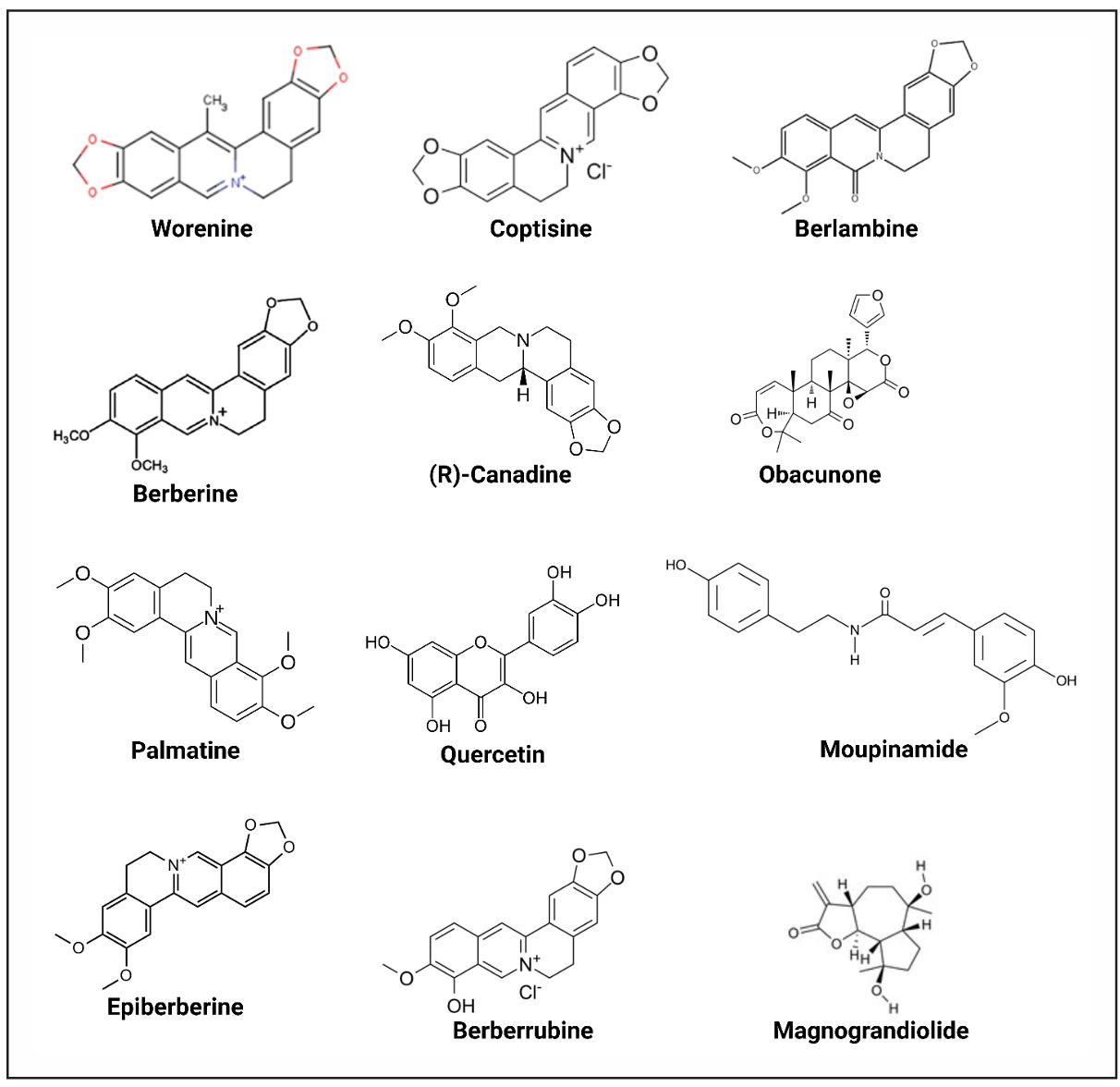
Figure 1. Chemical structures of the selected compounds. Illustrated the 2D chemical structures of the 12 CR-derived compounds (e.g., Worenine, Coptisine, Obacunone) identified in Table 1. Structures were generated using cheminformatics tools (e.g., SwissADME) and validated for consistency with PubChem entries.
Target collection for molecules
For the selected compounds, a total of 36,441 targets were identified where 5954 were duplicated and 30,487 targets were unique. From gene mapping we got 15,499 final targets for those molecules. The number of targets for each molecule is given in Table 2.
Table 2.
Selected molecules with identified targets.
| Compound | Targets | Compound | Targets |
|---|---|---|---|
| Worenine | 30,442 | Obacunone | 114 |
| Coptisine | 120 | Berberrubine | 105 |
| Berlambine | 101 | Palmatine | 137 |
| Epiberberine | 108 | Quercetin | 4574 |
| Berberine | 420 | Moupinamide | 104 |
| (R)-Canadine | 103 | Magnograndiolide | 100 |
Note: Quantifies the number of unique protein targets per compound, derived from CTD and Swiss Target Prediction databases. Quercetin had the highest target count (4,574), while Magnograndiolide had the lowest (100). Targets were mapped to Homo sapiens via UniProt.
Protein-protein interaction network
To establish molecular connections between Lewy body dementia and Coptidis Rhizoma compounds, the common target genes were identified. Among the collected 226 disease-related targets and 15,499 compound-associated targets, 214 common genes were found using the Venny tool (Figure 2A). These intersecting targets suggest a significant relationship between the herbal compounds and disease pathology. After that the genes of interest were subjected to insert in String for creating a PPI network. The network was constructed with 182 nodes and 606 edges (Figure 2B). By focusing on degree value of the nodes, we calculated the top 20 targets with the Cytohubba plugin. The top 20 hub genes are shown in Figure 2C. NGF, NEFL, MTOR, MAPT, ITGAM, GRN, GBA, APP, APOE, ANXA5, AIF1, TARDBP, SQSTM1, SNCA, SLC6A3, PSEN2, PRKN, PINK1, PRARK7, and NTRK1 are the identified key nodes (Figure 2D).
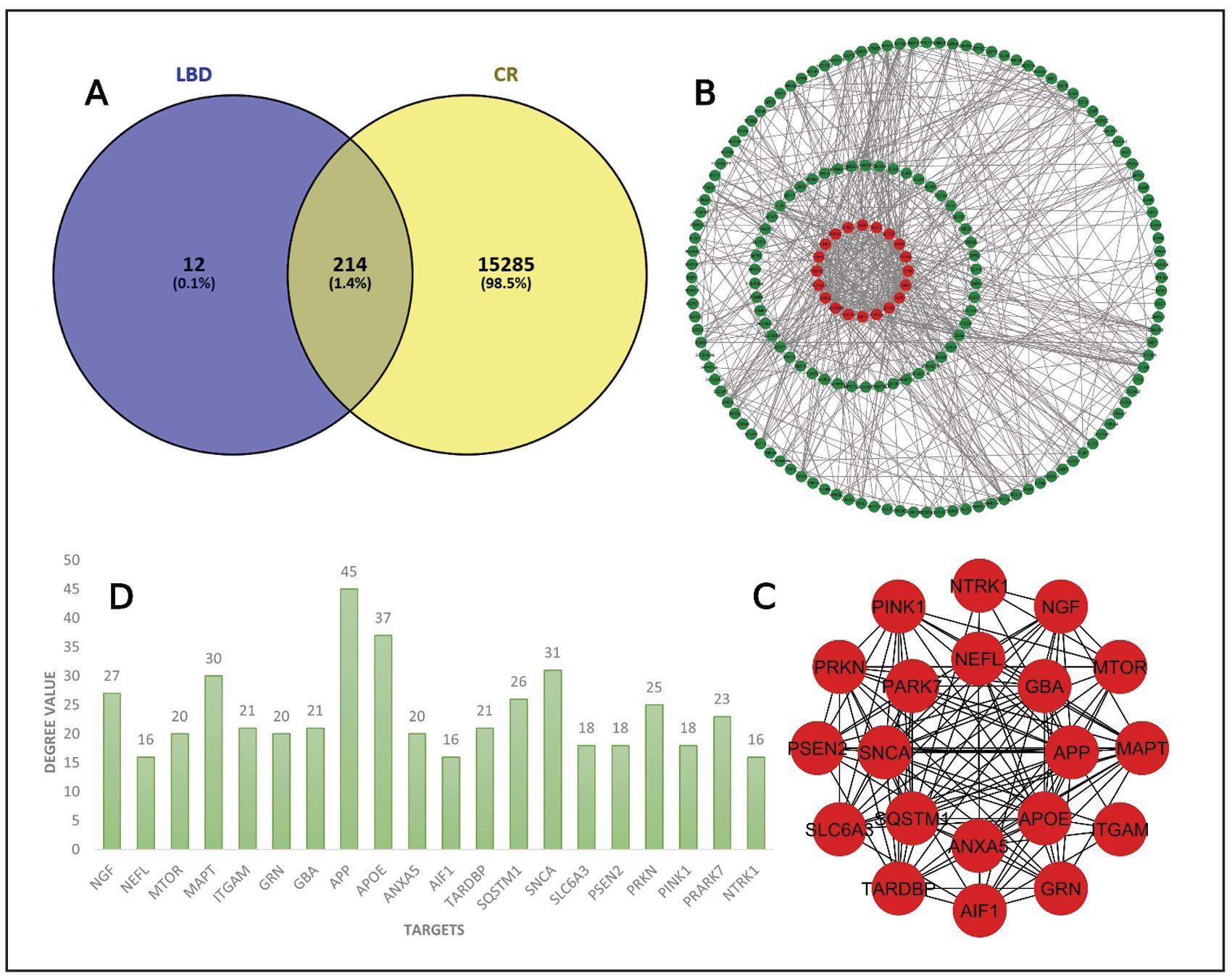
Figure 2. Network interactions analysis. (A) Intersect targets are showed in Venn diagram. (B) PPI interaction. (C) Interaction among top 20 hub genes. (D)Bar plots for top 20 hub genes with degree value.
Gene ontology (GO) with pathway analysis
To better understand the biological significance of the identified targets, we performed GO and KEGG pathway enrichment analysis using the DAVID platform. The analysis exhibited a total of 764 entries where Biological Process (BP) showed 212 results, Cellular components (CC) showed 213 results, 209 for Molecular functions (MF), and 130 results for KEGG pathway. BP terms displayed Negative regulation of neuron apoptotic process, adult locomotory behavior, cellular response to oxidative stress, microglial cell activation, dopamine uptake involved in synaptic transmission, neuron apoptotic process, regulation of mitochondrion organization, synapse organization, response to oxidative stress, and regulation of dopamine secretion (Figure 3A). Axon, membrane raft, dendrite, neuron projection, synaptic vesicle, lysosome, neuronal cell body, Golgi apparatus, growth cone, and lysosomal membrane were present in cellular components (Figure 3A). On the other hand, MF terms showed protein binding, ubiquitin protein ligase binding, identical protein binding, amyloid-beta binding, cuprous ion binding, protein-containing complex binding, enzyme binding, signaling receptor binding, structural molecule activity, and kinesin binding (Figure 3A).
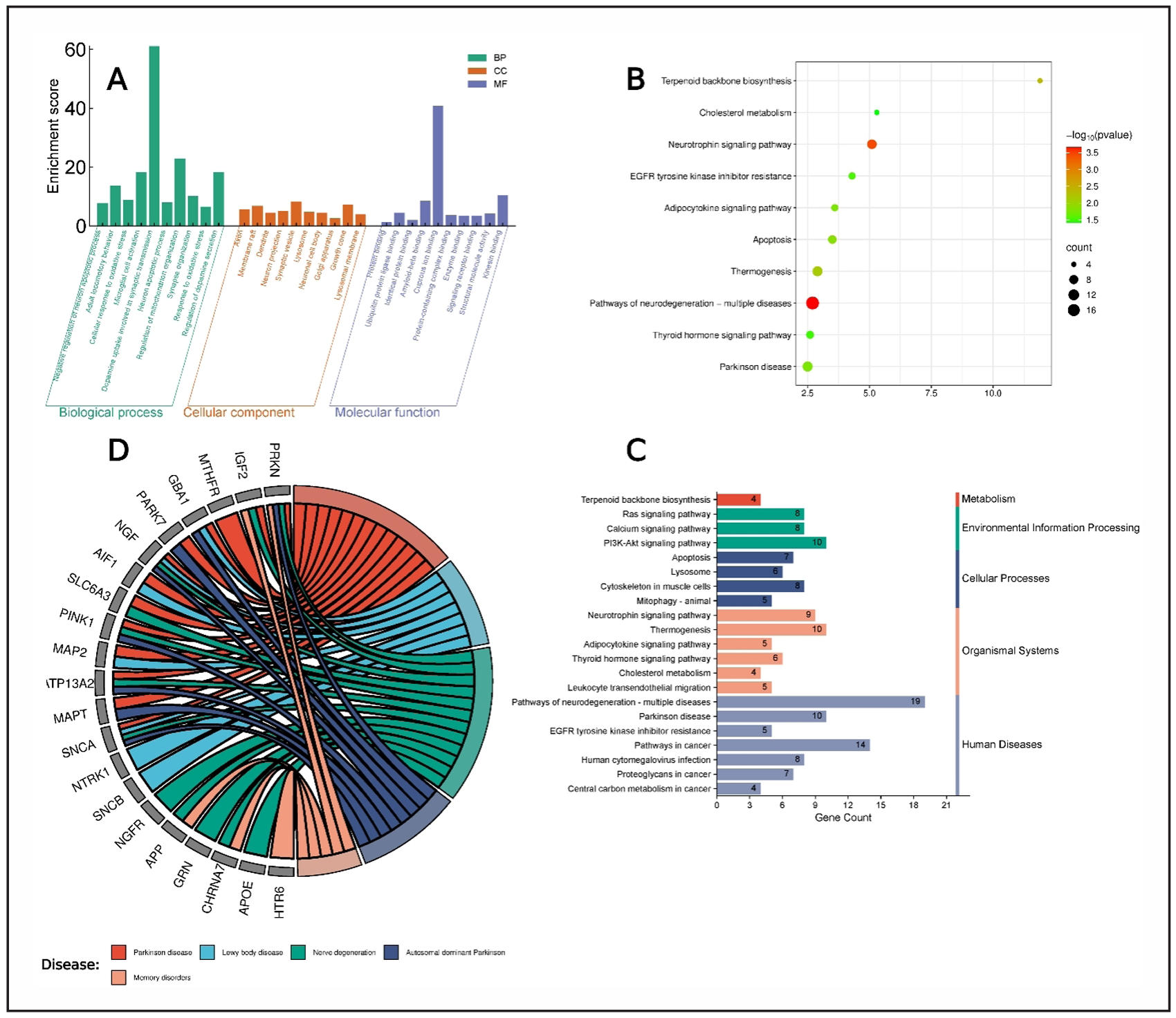
Figure 3. Gene Ontology and KEGG pathway analysis. (A) Different gene ontology functions present in bar chart. (B) KEGG enrichment bubble plot. (C) Different systems and actions of KEGG pathways. (D) Chord diagram linking targets to diseases.
Top 10 pathways were added in KEGG pathway enrichment plot based on P-value to visualize the most significant pathways related to disease where pathways of neurotrophin signaling pathway and pathways of neurodegeneration-multiple diseases are showing as highly significant pathways (Figure 3B). Furthermore, the 21 KEGG pathways based on p-value were selected for mapping. Those pathways were mapped on metabolism, environmental information processing, cellular process, organismal systems, and human diseases. The bars present that most of the pathways are involved in human diseases (Figure 3C). Additionally, we constructed a chord diagram based on targets related to different diseases to identify the associated diseases. The plot shows that Parkinson disease and Lewy body disease as the best results (Figure 3D). The colorful bubbles in the plot represents the -log10 (p-value) with a gradient from green to red. The red color indicates a higher -log10 (p-value) which is more significant, while green indicates a lower -log10 (p-value) which means less significant (Figure 3B).
MCODE analysis
The MCODE algorithm was utilized to find out the core therapeutic targets and based on this we constructed a modular network. Positive regulation of protein localization, regulation of neuron apoptotic process, behavior, inflammatory response, and response to oxidative stress are some of the functions from the topological analysis in Metascape (Figure 4A). To understand the relationship between pathways and targets, a subset network was constructed (Figure 4B). This network helps us to find out different important functions of targets within different clusters. Moreover, the Metascape performed MCODE cluster analysis on the 214 intersect genes. That analysis provided 5 target modules in treating Lewy body dementia by using those selected compounds (Figure 5). These modules help to get the biological pathways that might be present in therapeutic impacts on the disease.
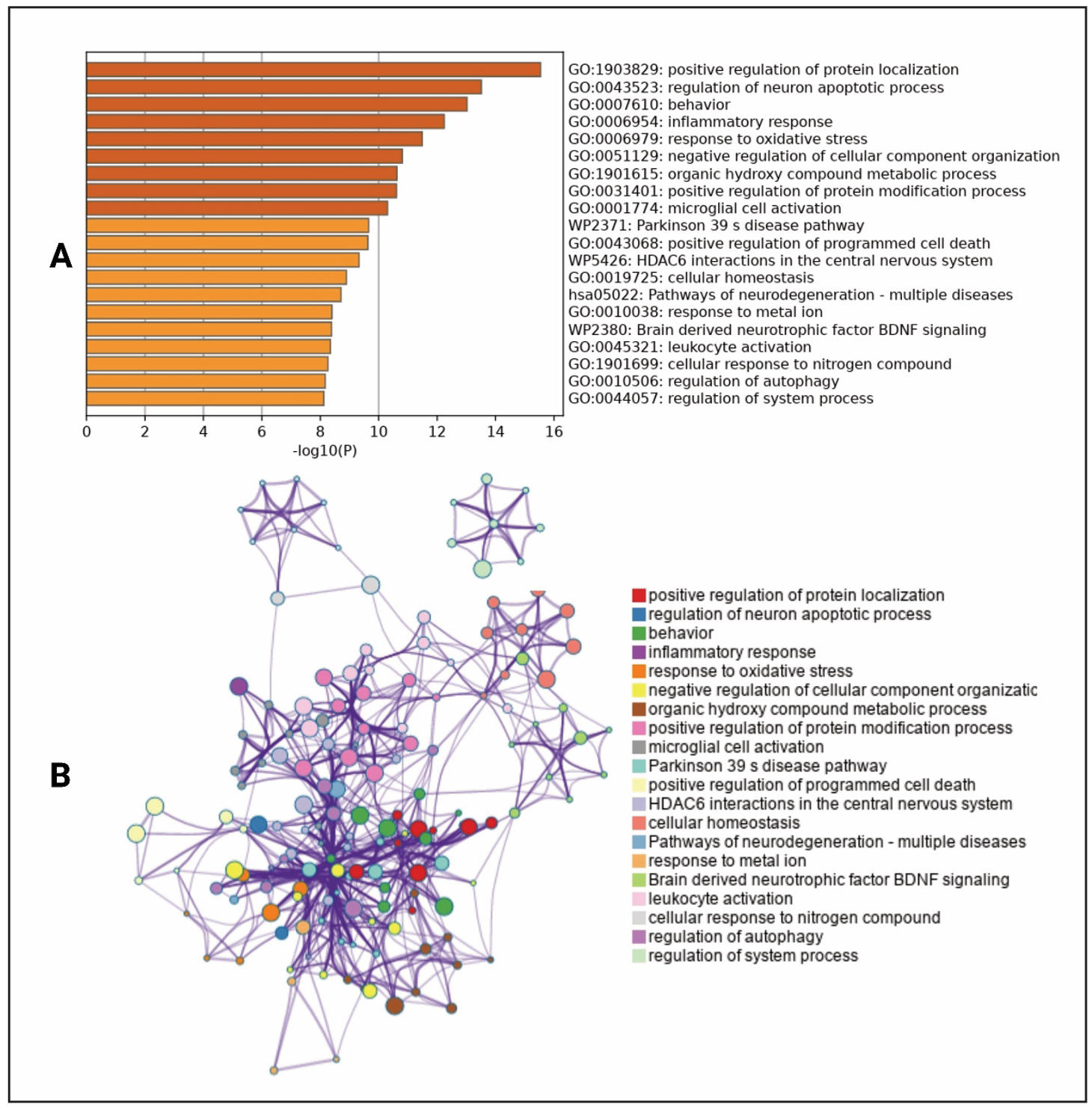
Figure 4. Expanded subset network of identified common targets enriched with high-significance terms. (A) Representation of high-significance enrichment terms. The colors in the bars represent the significance of different biological processes, pathways, or functions. Darker shades indicate higher significance levels based on the −log10(P) value, while lighter shades represent lower significance. This helps visually distinguish the most statistically relevant terms in the dataset. (B) Detailed subnetwork illustrating specific interactions within the identified target network.
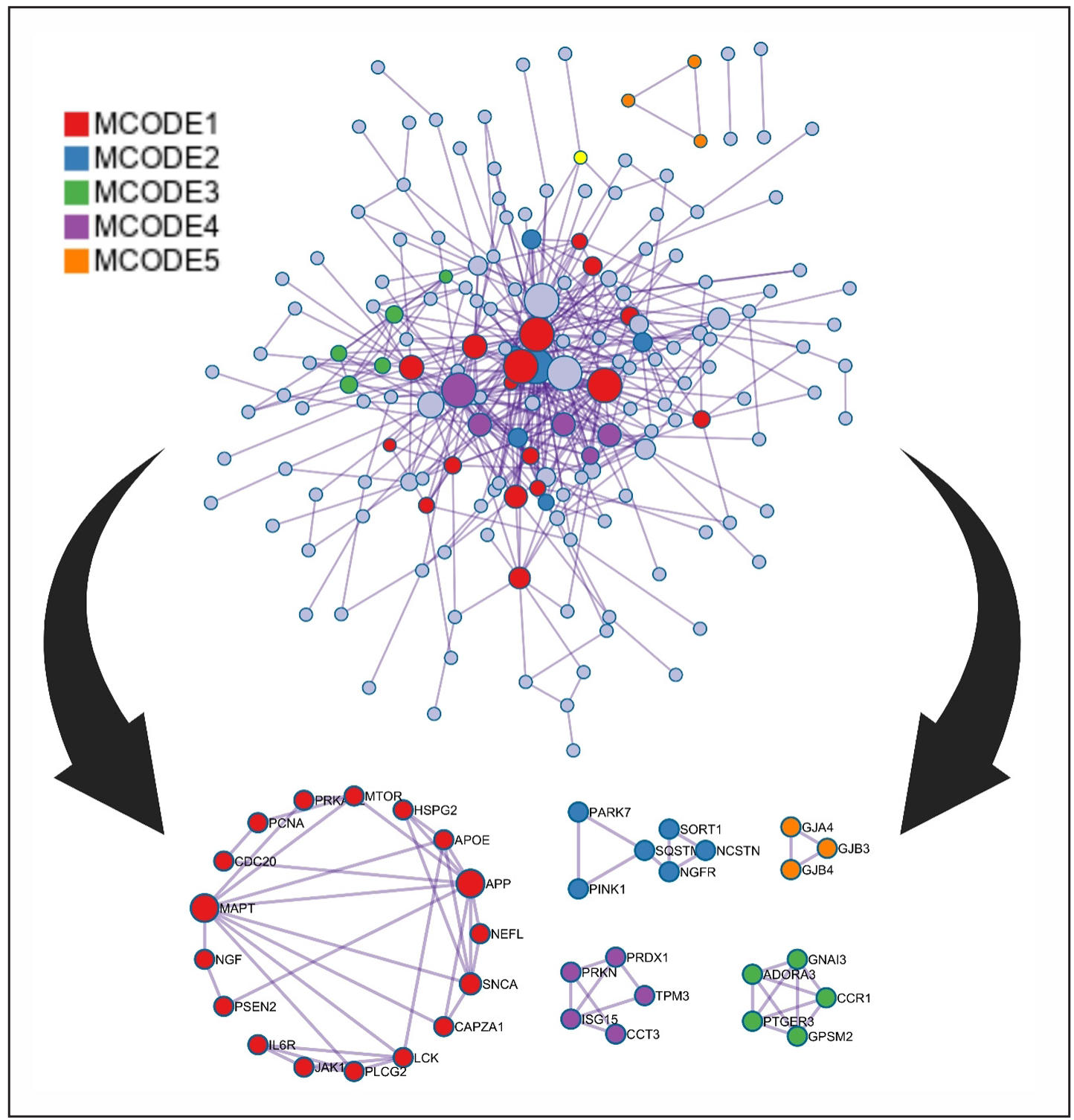
Figure 5. Protein-protein interaction (PPI) network enriched with comprehensive diagrammatic representations of distinct cluster modules.
Notably, these modules are enriched in different pathways, such as leukocyte activation, PID P75 NTR PATHWAY, G alpha (i) signaling events, HDAC6 interactions in the central nervous system, and Gap junction assembly (Table 3, Figure 5).
Table 3.
Pathway enrichment result for MCODE1, MCODE2, MCODE3, MCODE4, and MCODE5.
| Color | MCODE | GO | Description | Log10(P) |
|---|---|---|---|---|
| Red | MCODE_1 | GO:0045321 | leukocyte activation | -9.2 |
| Red | MCODE_1 | GO:1902903 | regulation of supramolecular fiber organization | -8.9 |
| Red | MCODE_1 | GO:0051129 | negative regulation of cellular component organization | -8.7 |
| Blue | MCODE_2 | M153 | PID P75 NTR PATHWAY | -9.5 |
| Blue | MCODE_2 | R-HSA-205043 | NRIF signals cell death from the nucleus | -9.0 |
| Blue | MCODE_2 | R-HSA-204998 | Cell death signaling via NRAGE, NRIF and NADE | -6.9 |
| Green | MCODE_3 | R-HSA-418594 | G alpha (i) signaling events | -7.4 |
| Green | MCODE_3 | R-HSA-388396 | GPCR downstream signaling | -6.2 |
| Green | MCODE_3 | R-HSA-372790 | Signaling by GPCR | -6.0 |
| Blue | MCODE_4 | WP5426 | HDAC6 interactions in the central nervous system | -6.3 |
| Orange | MCODE_5 | R-HAS-190861 | Gap junction assembly | -8.8 |
| Orange | MCODE_5 | R-HAS-190828 | Gap junction trafficking | -8.6 |
| Orange | MCODE_5 | R-HAS-157858 | Gap junction trafficking and regulation | -8.5 |
Target-pathway network
To visualize the connections between identified targets and key biological pathways, a target-pathway interaction network was constructed by using Cytoscape 3.10.3. The network consisted of 208 nodes and 667 edges (Figure 6A), allowing the identification of the top 20 hub genes with significant pathway involvement. The deep blue circle shape represents the pathways and red circle shapes indicating target components. After that a network constructed among the top 20 targets based on their degree value by using cytohubba plugin in Cytoscape (Figure 6B) and their degree values are present in bar chart (Figure 6C). PINK1, PARK7, NTRK1, NRAS, NGF, MTOR, TARDBP, SQSTM1, SNCA, PSEN2, PRKN, MAPT, ITGAM, GBA, APP, APOE, MFN2, SLC6A3, ANXA5, and GRN are the top 20 hub genes from the target-pathway network. To get the common targets, the top 20 targets from protein-protein interaction and the top 20 targets from target-pathway network interaction were used to intersect and visualized in Venn diagram (Figure 6D). Finally, NGF, MTOR, MAPT, ITGAM, GRN, GBA, APP, APOE, ANXA5, TARDBP, SQSTM1, SNCA, SLC6A3, PSEN2, PRKN, PINK1, PARK7, and NTRK1 were identified as significant targets.
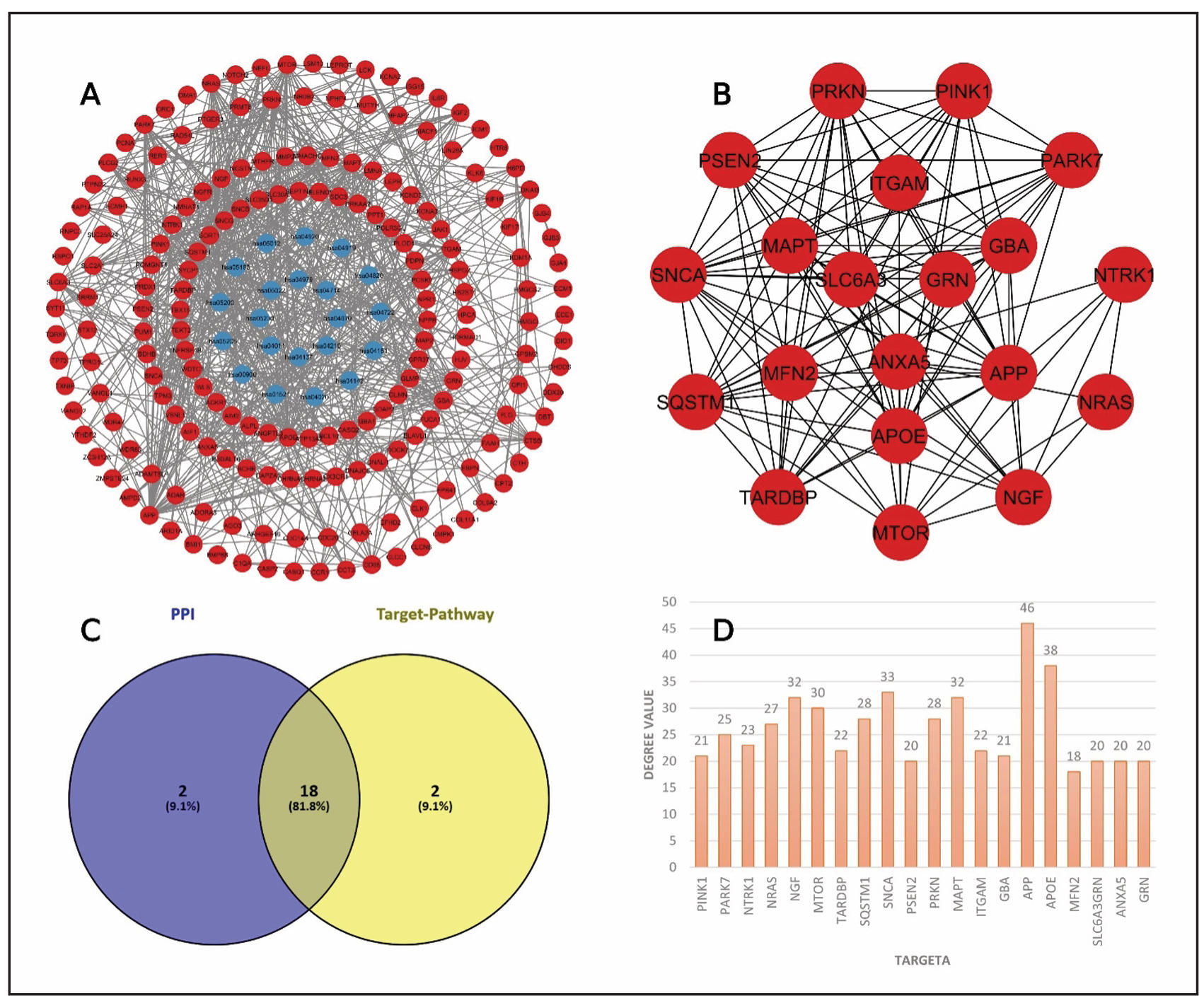
Figure 6. Target-pathway network interaction and common targets identification. (A)Pathway-target network diagram. (B) PPI network among top 20 targets. (C) Similar target identification between PPI network and target-pathway network. (D) Bar chart for top 20 genes with degree value.
Key targets selection
The PPI network from String database was subjected to use in Cytoscape software to get top ten genes for the five different topological properties Maximal Clique Centrality (MCC), Density of Maximum Neighborhood Component (DMNC), Bottle-Neck, Closeness, and Degree value. We created a simple network of the common 18 targets from PPI network and target-pathway network to visualize their interactions (Figure 7A). A Venn diagram among the five topological properties was constructed to identify the common targets (Figure 7B). GBA, MAPT, PRKN, SNCA, and TARDBP are the common targets among those properties.
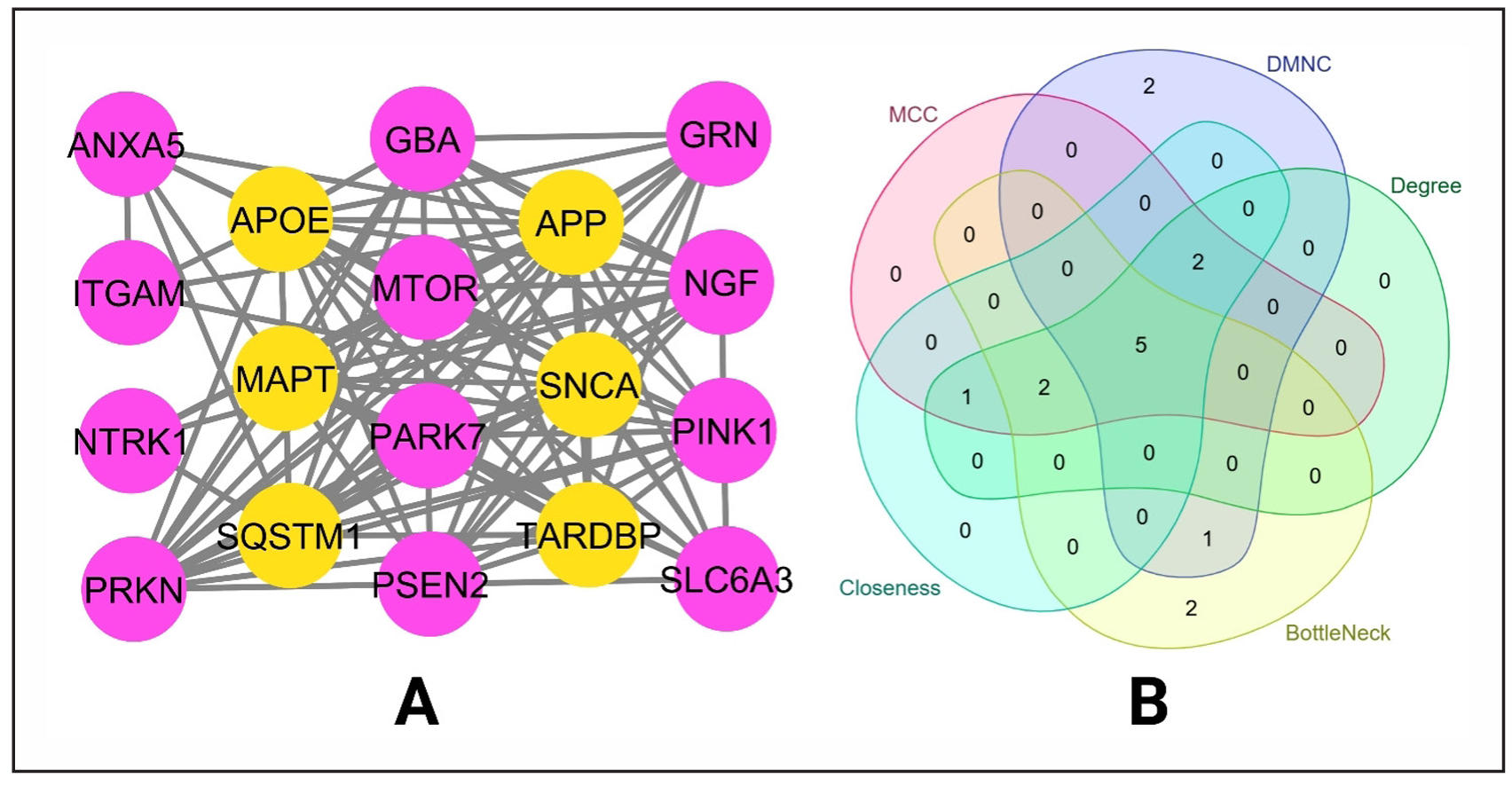
Figure 7. Identification of key targets based on different properties. (A) Constructed network between the 18 hub genes. (B) Identifying common targets based on topological properties.
The analysis of the two figures reveals significant enrichment of pathways and
gene ontology (GO) terms associated with neurodegenerative diseases and neuronal
function. In Figure 8A, KEGG pathway enrichment highlights key neurodegenerative
conditions, including Parkinson's disease, Alzheimer's disease, amyotrophic lateral
sclerosis (ALS), and mitophagy, with highly significant false discovery rates (FDRs)
ranging from 1.7e-06 to 1.0e-03. The neurotrophin signaling pathway was also
prominent, suggesting a role in neuronal survival and maintenance. Additionally,
biological process GO terms such as regulation of neuron apoptosis, dopamine
transport, and microglial cell activation were enriched, with extremely low FDRs
(as stringent as 1.0e-13), underscoring their importance in neuronal health and
degeneration (Figure 8B).
Figure 9 further supports these findings by identifying enriched cellular
components and molecular functions. Cellular structures like Lewy bodies—a
hallmark of Parkinson’s disease—along with neuronal cell bodies, axons, and
glial projections were significantly enriched (Figure 9A). Molecular function
analysis revealed critical roles for protein-binding activities, including
chaperone binding (e.g., heat shock proteins), neurotrophin receptor binding,
and ubiquitin-related processes, all of which are implicated in protein misfolding
and degradation pathways (Figure 9B). These results collectively emphasize disruptions
in protein homeostasis, mitochondrial function, and neuronal survival mechanisms,
providing a molecular framework for understanding neurodegeneration. The consistent
involvement of these pathways suggests potential therapeutic targets for mitigating
neuronal damage in related disorders.
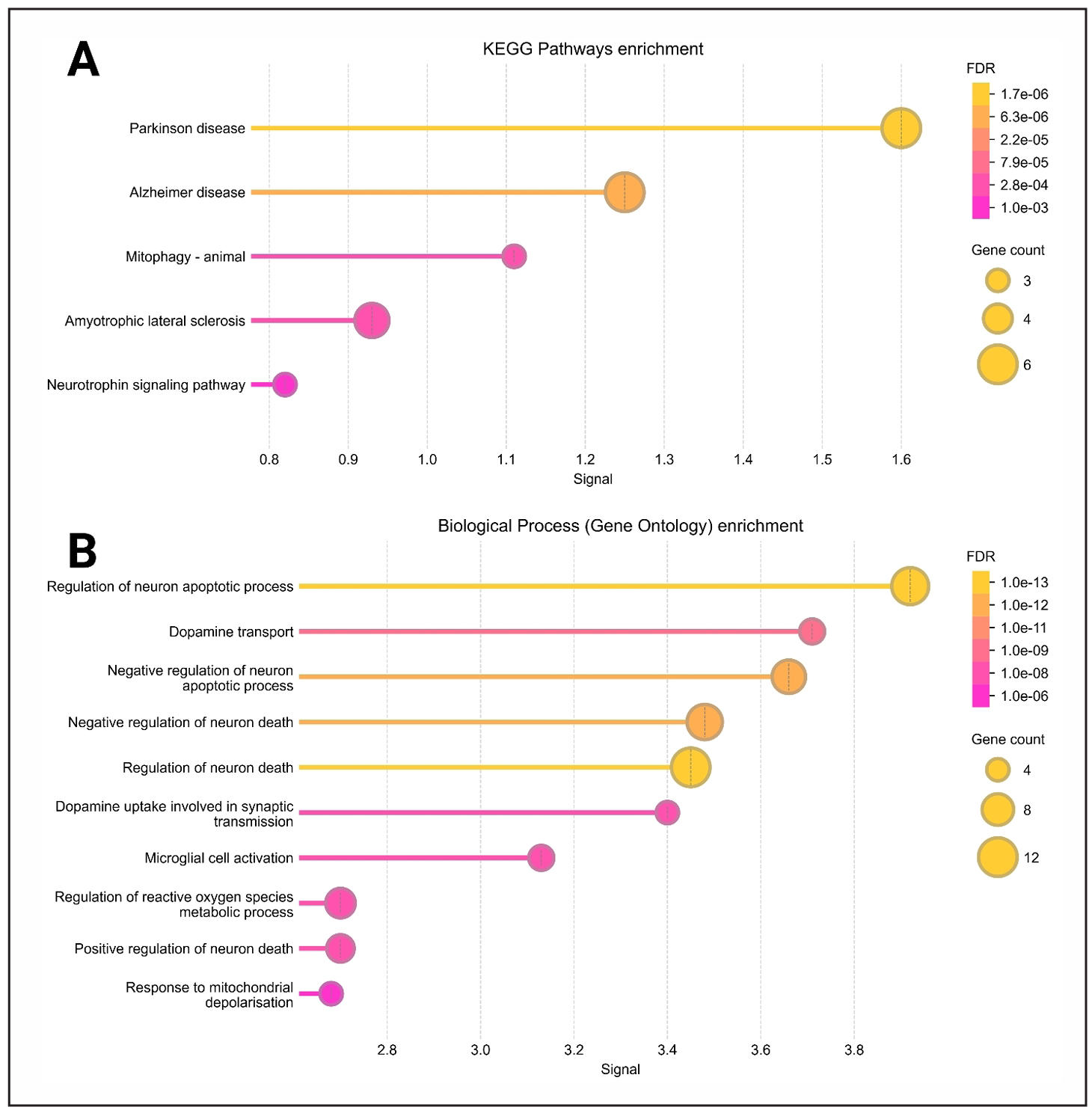
Figure 8. Gene ontology enrichment analysis for cellular components and molecular functions based on the 18 common hub genes. This figure illustrates the enrichment of gene ontology terms, with panel A focusing on cellular components and panel B highlighting molecular functions.
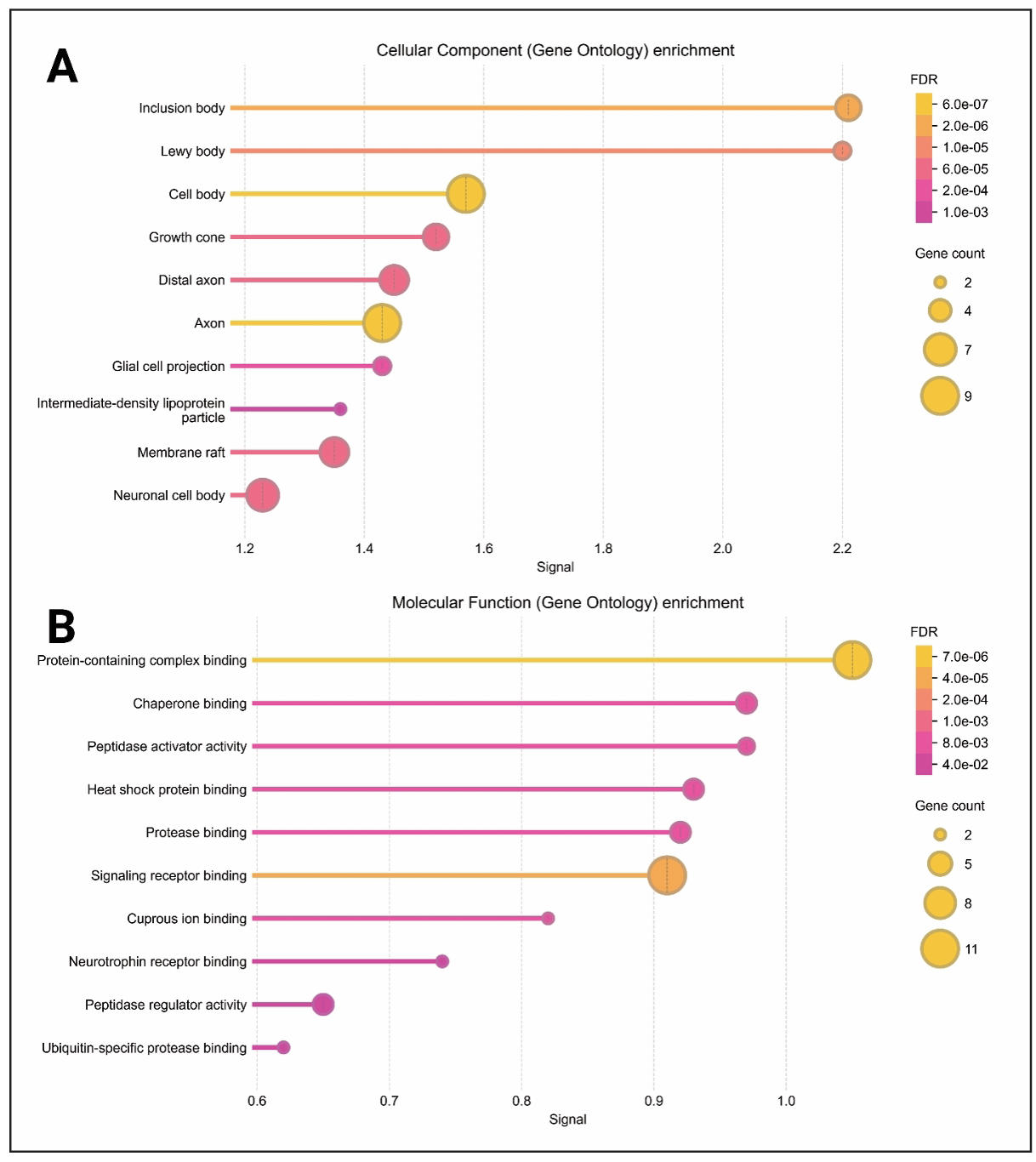
Figure 9. KEGG pathway and gene ontology biological process enrichment analysis based on the 18 common hub genes. This figure presents enrichment analysis results, highlighting KEGG pathways (A) and Gene Ontology biological processes (B).
Docking result
Molecular docking was employed to assess the binding interactions between selected bioactive compounds and key proteins implicated in Lewy body dementia. The docking results provided valuable insights into ligand-protein affinity, where compounds such as Obacunone and Coptisine demonstrated strong interactions, reinforcing their potential for therapeutic applications (Figure 10-14). The lower the binding affinity means the stronger interaction between a compound and protein. The binding results of the targets and compounds were summarized in Table 4 and most of the affinities were less than -5 kcal/mol. Usually, the binding value less than -7 kcal/mol means strong predicted binding, value less than -5 kcal/mol suggests moderate binding [52].
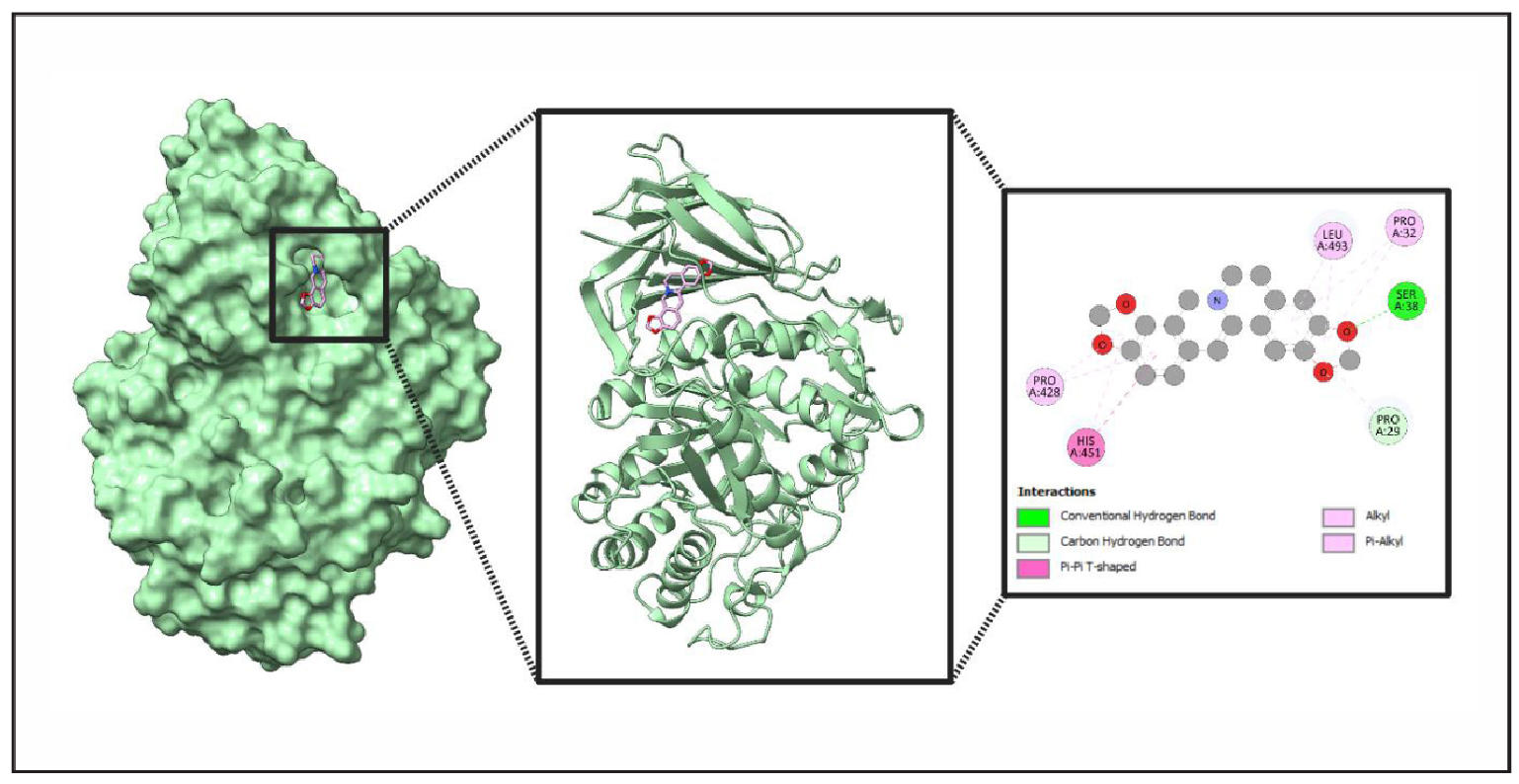
Figure 10. Comprehensive Structural and Interaction Analysis of Ligand (Coptisine) Binding to Protein (GBA). This figure presents a multi-layered examination of ligand binding within a protein’s active site. The leftmost panel visualizes the protein’s surface representation, highlighting the ligand within the active site. The middle panel showcases the ribbon model, emphasizing secondary structure elements surrounding the ligand. The rightmost panel provides a molecular interaction map, detailing conventional hydrogen bonds, carbon hydrogen bonds, Pi-Pi T-shaped interactions, alkyl interactions, and Pi-alkyl interactions. Key amino acids engaged in these interactions include LEU A:493, SER A:38, PRO A:32, PRO A:29, PRO A:428, and HIS A:451.
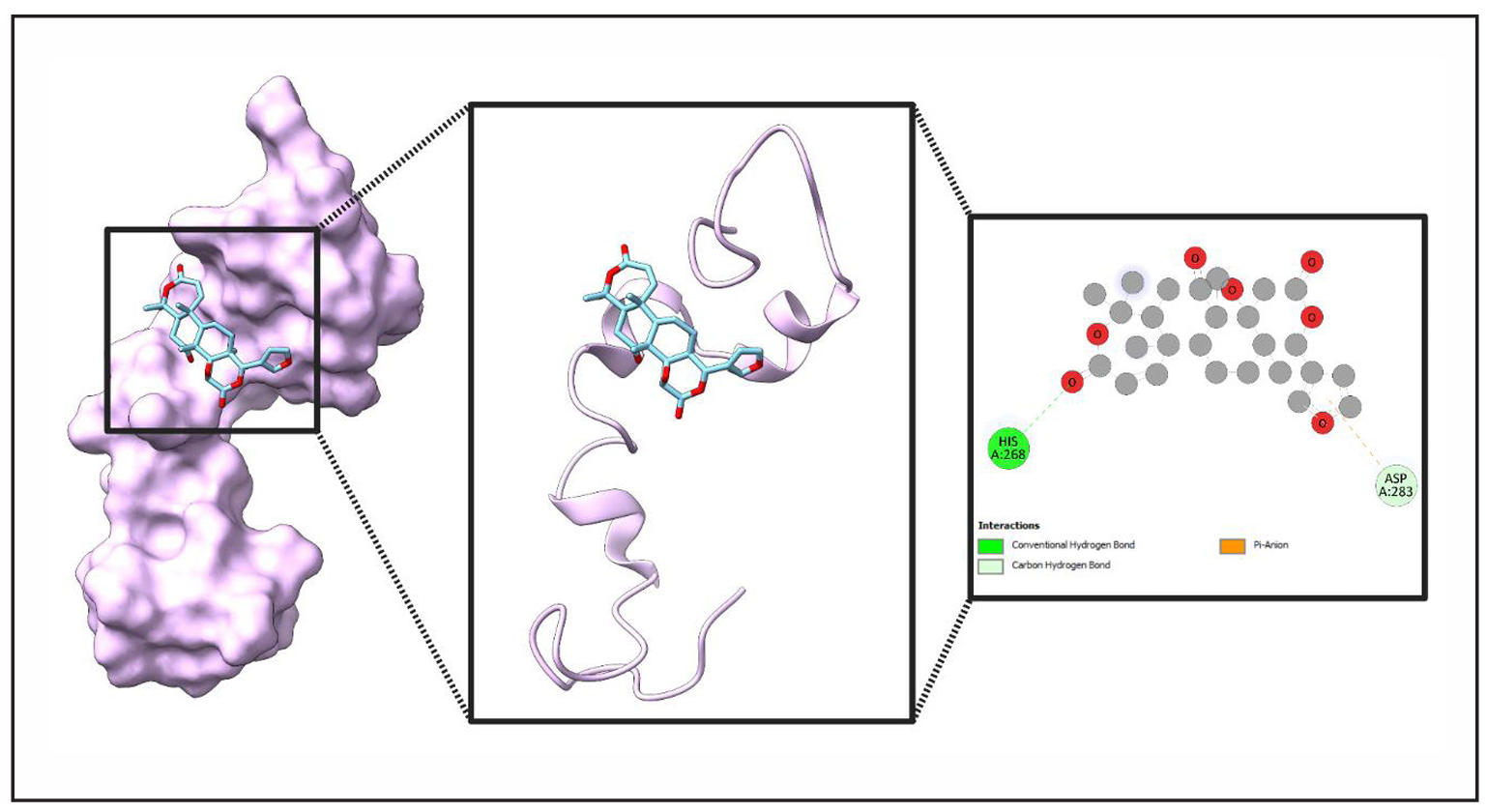
Figure 11. Molecular Interaction of a Ligand (Obacunone) with Protein’s (MAPT) binding site. This figure provides an in-depth visualization of the ligand binding to a protein's active site. The leftmost panel illustrates the ligand embedded within the protein’s surface, represented in purple. The middle panel showcases the ribbon model, highlighting secondary structure elements surrounding the ligand, with atomic colors (carbon in light blue, oxygen in red, nitrogen in blue) reflecting molecular composition. The rightmost panel presents a detailed interaction map, depicting conventional hydrogen bonds (green), carbon hydrogen bonds (white), and pi-anion interactions (orange). Key interacting residues include HIS A:268 and ASP A:283, which play a crucial role in ligand stabilization.
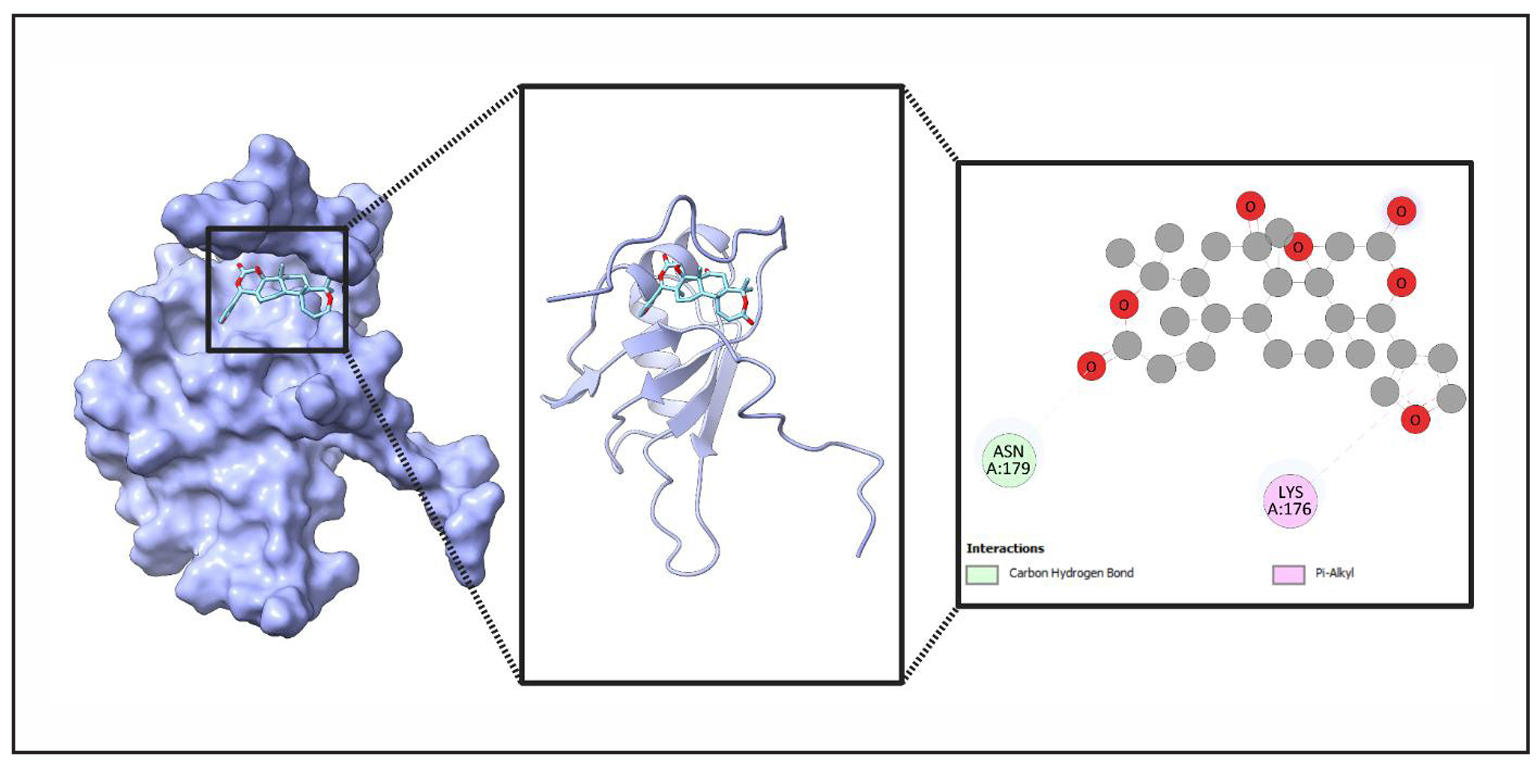
Figure 12. Detailed Structural Analysis of Ligand (Obacunone) Binding to Protein (TARDBP). This figure provides a comprehensive visualization of a ligand binding to a protein’s active site. The leftmost panel displays the protein’s surface representation with the ligand nestled in the binding pocket. The middle panel offers a ribbon model view, emphasizing secondary structure elements surrounding the ligand. The rightmost panel presents an interaction map, detailing carbon hydrogen bonds and Pi-alkyl interactions with specific amino acids, ASN A:179 and LYS A:176.
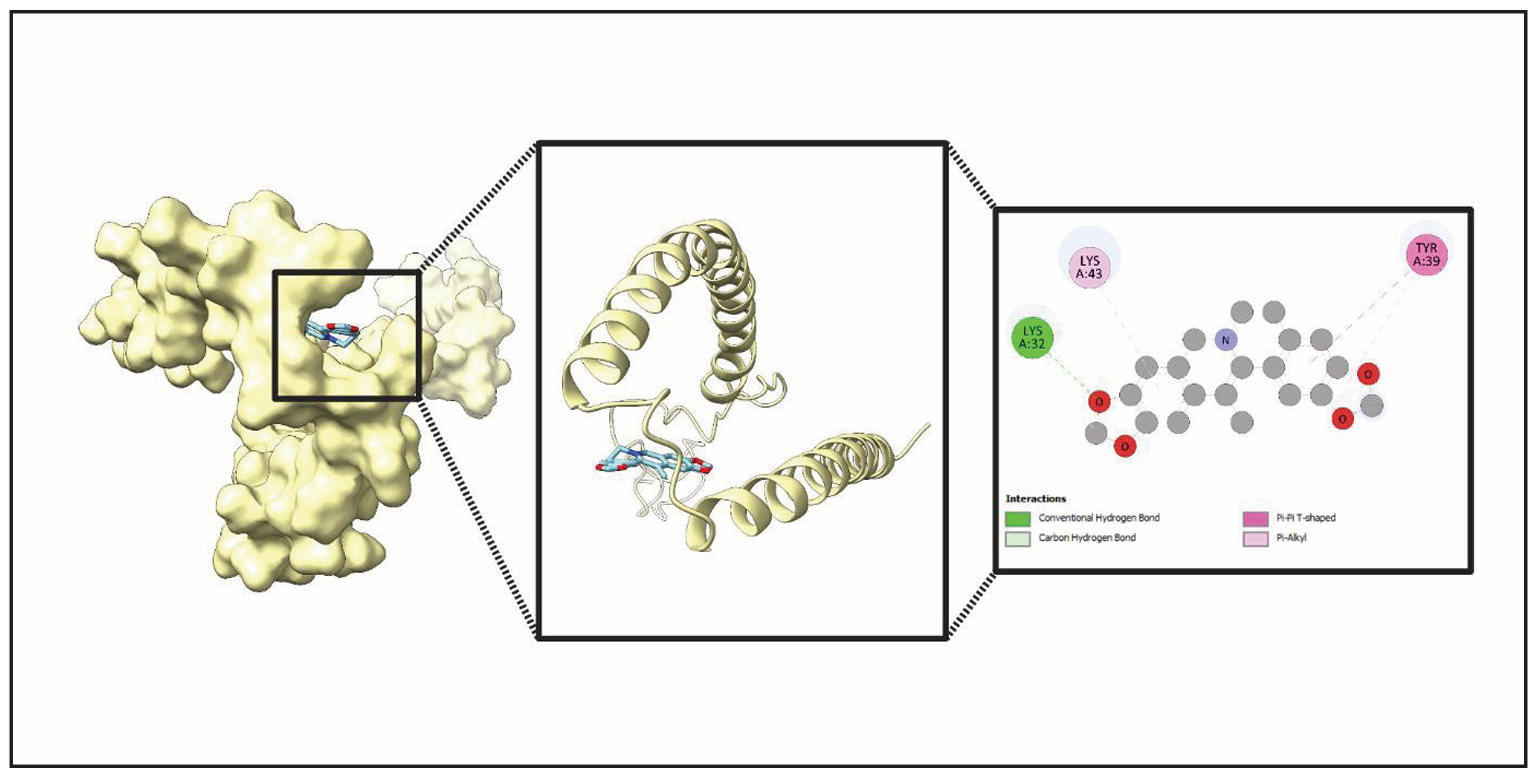
Figure 13. Detailed structural analysis of ligand (Worenine) binding with a Protein (SNCA). This figure presents an in-depth examination of the ligand binding within the protein’s active site. The leftmost panel illustrates the protein’s surface representation, showing the ligand positioned within the binding pocket. The middle panel highlights the ribbon model, emphasizing secondary structure elements surrounding the ligand. The rightmost panel provides a molecular interaction map, detailing conventional hydrogen bonds, carbon hydrogen bonds, Pi-Pi T-shaped interactions, and Pi-alkyl interactions. Key residues involved include Lysine (Lys) at positions A:32 and A:43, as well as Tyrosine (Tyr) at position A:39.
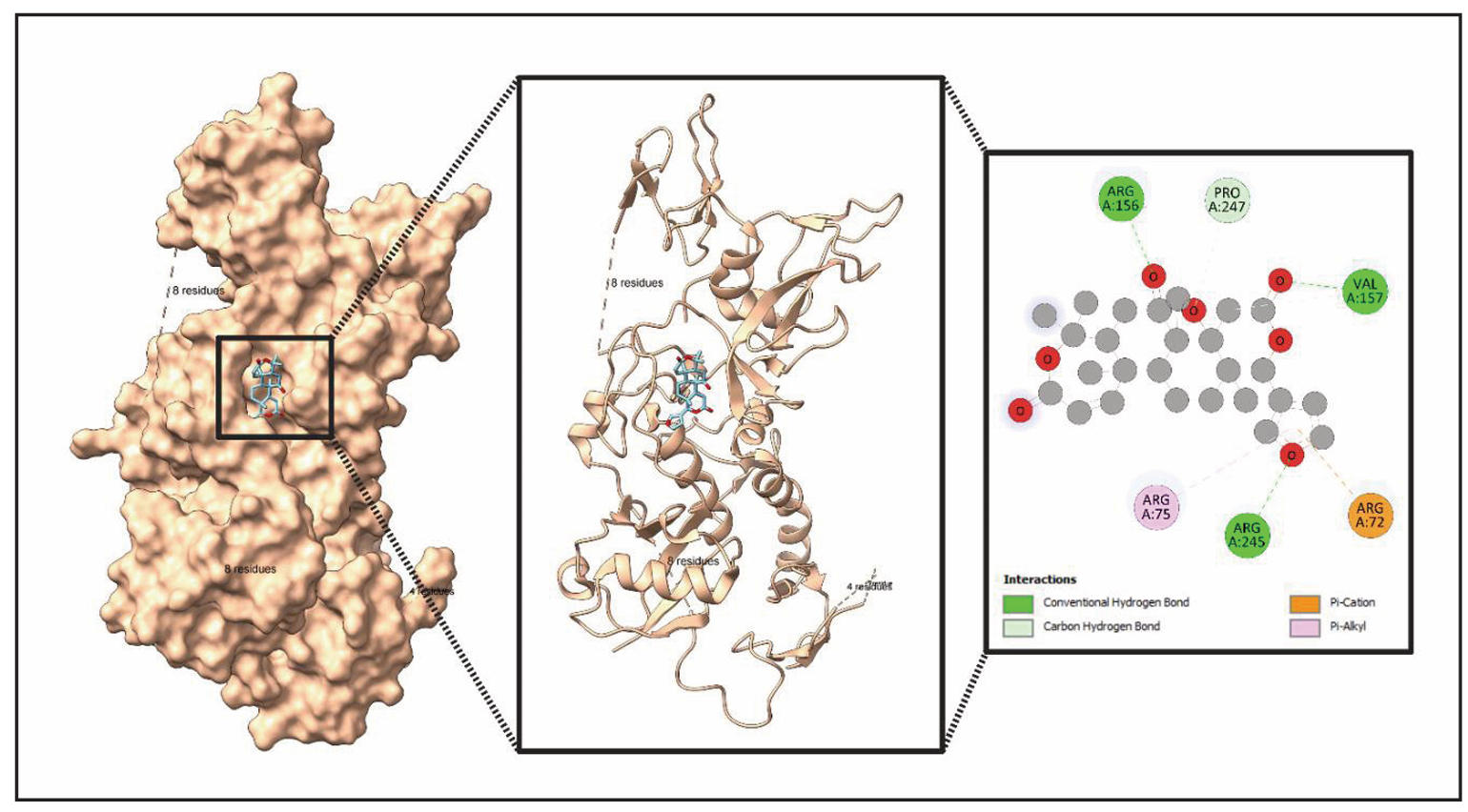
Figure 14. Structural analysis of ligand (Obacunone) binding in protein (PRKN). This figure presents a detailed structural examination of a ligand binding within a protein's active site. The left panel displays the protein’s surface representation, showing the ligand positioned in the binding pocket. The middle panel highlights the secondary structure of the protein, emphasizing helices and the ligand's placement. The right panel provides a molecular interaction map, detailing key residues—Lysine (Lys) A:32, Lysine (Lys) A:43, and Tyrosine (Tyr) A:39—and the types of interactions, including conventional hydrogen bonds, carbon hydrogen bonds, Pi-Pi T-shaped interactions, and Pi-alkyl interactions.
Table 4.
Binding scores of different targets with different molecule.
| Compound name | Compound CID | Protein name | ||||
|---|---|---|---|---|---|---|
| GBA | MAPT | TARDBP | SNCA | PRKN | ||
| Binding affinity (kcal/mol) | ||||||
| Worenine | 20055073 | -9.3 | -6.6 | -6.2 | -7.4 | -8.9 |
| Coptisine | 72322 | -9.4 | -6.2 | -6.0 | -7.1 | -8.7 |
| Berlambine | 11066 | -8.8 | -5.9 | -5.9 | -6.7 | -8.3 |
| Epiberberine | 160876 | -8.7 | -5.7 | -5.7 | -6.4 | -7.8 |
| Berberine | 2353 | -8.8 | -5.8 | -5.5 | -6.4 | -7.8 |
| (R)-Canadine | 443422 | -8.0 | -5.8 | -5.9 | -6.4 | -7.7 |
| Obacunone | 119041 | -9.4 | -7.0 | -6.4 | -7.1 | -9.2 |
| Berberrubine | 72704 | -8.7 | -5.9 | -5.9 | -6.6 | -8.2 |
| Palmatine | 19009 | -8.2 | -5.4 | -5.1 | -6.0 | -7.4 |
| Quercetin | 5280343 | -8.5 | -5.9 | -5.6 | -6.3 | -8.0 |
| Moupinamide | 5280537 | -7.7 | -6.2 | -5.8 | -5.8 | -7.6 |
| Magnograndiolide | 5319198 | -8.1 | -5.2 | -5.5 | -6.0 | -7.4 |
The molecular docking results presented compelling evidence for the therapeutic
potential of various compounds, particularly regarding their interactions with
five key target proteins: GBA, MAPT, TARDBP, SNCA, and PRKN. The docking scores,
which indicate binding affinities, suggest that certain compounds exhibit strong
potential for protein-ligand interactions (Table 4, Figure 15). Among them,
Obacunone demonstrated the highest binding affinities, showing significant
interactions with GBA (-9.4 kcal/mol), MAPT (-7.0 kcal/mol), TARDBP (-6.4 kcal/mol),
and PRKN (-9.2 kcal/mol). These consistently strong binding affinities suggest that
Obacunone may be particularly effective in engaging multiple biological targets,
which could make it a promising candidate for further study, particularly in cases
where these proteins play critical roles in disease mechanisms.
Additionally, Coptisine and Worenine exhibited notable binding affinities,
reinforcing their potential in molecular interaction studies (Table 4, Figure 15).
Coptisine showed a high affinity for GBA (-9.4 kcal/mol), comparable to Obacunone,
suggesting that its molecular structure is well-suited for interacting with this
protein’s binding site. Meanwhile, Worenine exhibited the strongest interaction
with SNCA (-7.4 kcal/mol), an observation that could be particularly relevant
for studying neurodegenerative disorders such as Parkinson’s disease, given SNCA’s
role in disease pathology. These findings suggest that both Coptisine and Worenine
warrant further exploration to determine their specific mechanisms of binding and
biological relevance.
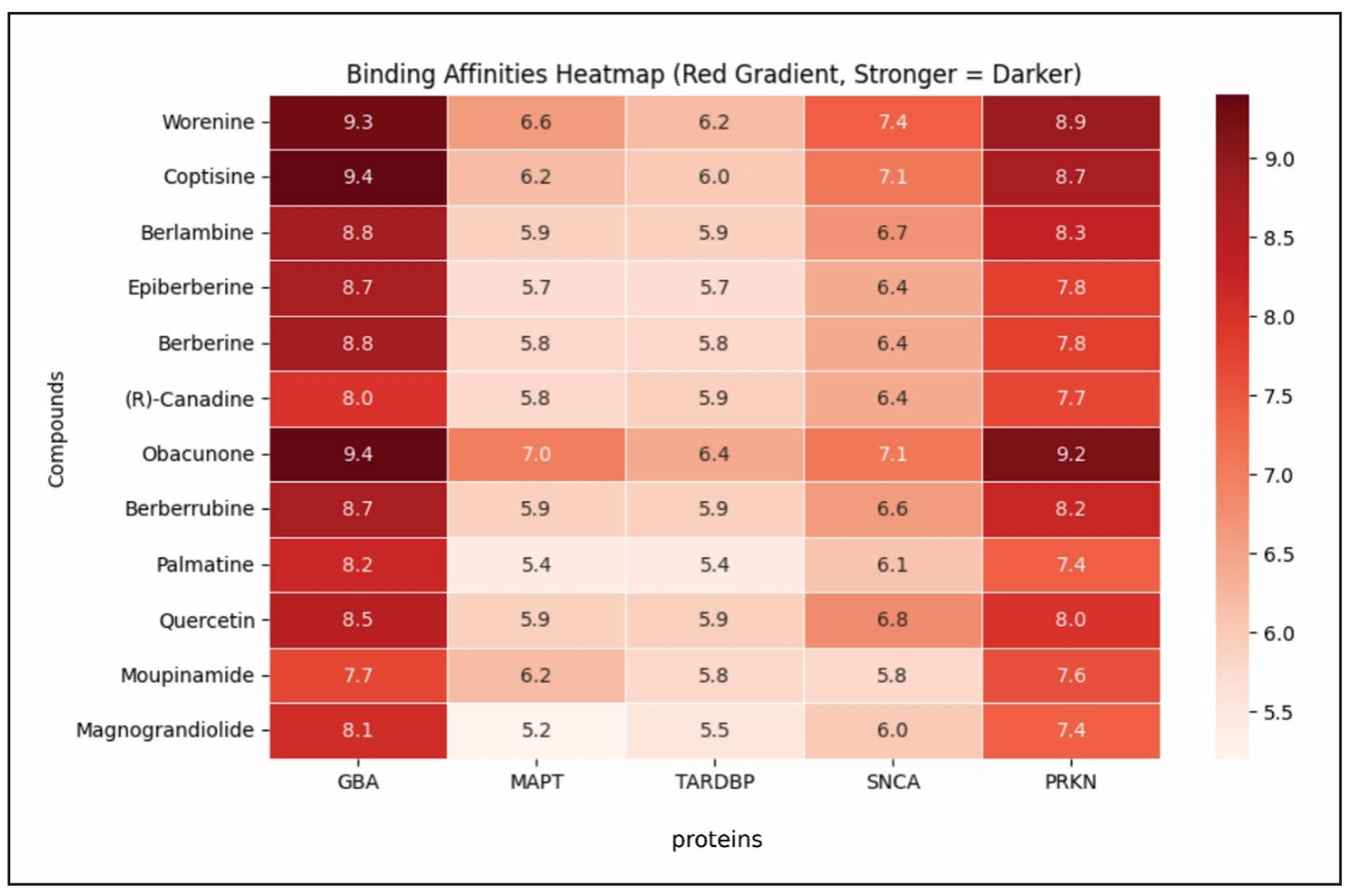
Figure 15. Heatmap of molecular docking-based binding affinities (in kcal/mol) between natural compounds and Lewy body dementia associated protein targets (GBA, MAPT, TARDBP, SNCA, PRKN). Red gradient indicates stronger binding (Higher Affinity = Darker).
In contrast, Magnograndiolide and Moupinamide exhibited weaker affinities
across multiple proteins. Magnograndiolide recorded the weakest interactions
with MAPT (-5.2 kcal/mol) and TARDBP (-5.5 kcal/mol), indicating relatively
weak engagement with these targets. Similarly, Moupinamide showed the lowest
binding affinity for SNCA (-5.8 kcal/mol), suggesting limited interaction stability
with this protein (Table 4, Figure 15). These higher binding energy values indicate
reduced molecular interaction, potentially limiting the therapeutic effectiveness
of these compounds unless structural modifications are made to enhance their affinity.
When assessing overall trends across the docking data, GBA exhibited the strongest
average affinity (-8.6 kcal/mol), followed closely by PRKN (-8.0 kcal/mol),
suggesting that these proteins may have well-defined active sites conducive to
stable ligand binding. Conversely, MAPT and TARDBP displayed the weakest average
binding affinities (approximately -5.9 kcal/mol each).
Discussion
Our computational study provides valuable insights into the therapeutic
potential of Coptidis Rhizoma (CR) in treating Lewy body dementia (LBD).
By integrating bioinformatics approaches, we explored the molecular interactions
between CR compounds and LBD-associated targets, unveiling a complex network of
therapeutic mechanisms. Through protein-protein interaction (PPI) network analysis,
we identified 20 critical hub genes, including TARDBP, MAPT, GBA, SNCA, and PRKN,
which play essential roles in neurodegenerative processes. MAPT stabilizes
microtubules, though its aggregation forms neurofibrillary tangles, contributing
to neurodegeneration [53, 54]. GBA mutations lead to lysosomal dysfunction,
promoting α-synuclein accumulation and Lewy body pathology [55-57]. SNCA
aggregates interfere with synaptic transmission and neuronal integrity,
exacerbating neurodegenerative decline [58, 59]. PRKN regulates mitochondrial
homeostasis and mitophagy, with dysfunction impairing cellular clearance
mechanisms [60-62]. Additionally, TARDBP is involved in RNA processing and
stress granule formation, with its mis-localization leading to neuronal
dysfunction and inclusion body formation, a hallmark of several neurodegenerative
diseases [63-65]. These targets highlight key molecular drivers of Lewy body
dementia (LBD), suggesting that Coptidis Rhizoma (CR) compounds may modulate
neuroprotective pathways and offer promising therapeutic interventions.
The pathway analysis revealed that CR compounds significantly impact neurotrophic
signaling, oxidative stress response, synaptic transmission, and mitophagy regulation,
all of which are integral to LBD pathology. The neurotrophic signaling pathway
supports neuronal survival and regeneration [66, 67], while oxidative stress
regulation is crucial in mitigating neurodegenerative damage [68, 69]. Synaptic
transmission stabilization, particularly involving SNCA and dopaminergic pathways,
suggests a possible improvement in cognitive function and motor coordination [70, 71].
Additionally, CR compounds may enhance mitophagy and autophagy regulation,
particularly through interactions with PRKN, which is essential in maintaining
mitochondrial integrity. Together, these findings indicate a multi-target
therapeutic mechanism where CR compounds could simultaneously restore neurotrophic
support, reduce oxidative damage, and enhance neuronal clearance processes.
For a proper molecular docking, the active site of each protein was identified
using Discovery Studio [72], a widely recognized computational tool known for its
robust structural analysis capabilities [73-75]. Unlike online tools that
merely confirm the presence of an active site, Discovery Studio allows a more
refined approach, enabling precise characterization based on ligand interactions,
binding pocket geometry, and energetic considerations [76, 77]. This level of
structural validation strengthens the reliability of the identified active sites,
ensuring consistency with established biochemical principles.
Molecular docking was utilized to evaluate the binding interactions between
bioactive compounds from Coptidis Rhizoma (CR) and key proteins implicated in
Lewy body dementia (LBD). The findings highlight strong affinity between
several compounds and LBD-associated targets, suggesting potential therapeutic
applications. Among the tested molecules, Obacunone exhibited the highest
binding affinity, particularly interacting with GBA (-9.4 kcal/mol) and
PRKN (-9.2 kcal/mol). This suggests a strong likelihood of Obacunone modulating
the biological function of these proteins, potentially contributing to neuroprotection.
Similarly, Coptisine demonstrated notable interaction with GBA (-9.4 kcal/mol),
reinforcing its therapeutic relevance. The robust binding affinities of these
compounds indicate promising potential for further investigation. On the other
hand, compounds like Magnograndiolide and Moupinamide displayed weaker binding,
particularly for MAPT and TARDBP, with docking scores around -5 kcal/mol.
Such interactions suggest a lower likelihood of significant biological engagement,
although structural modifications may enhance their efficacy.
Our research builds upon existing knowledge by bridging traditional herbal
medicine with advanced computational methodologies. The identification of
close associations with Parkinson's disease and Lewy body dementia pathways
highlights the interconnected nature of neurodegenerative disorders. The modular
network analysis further demonstrated five distinct target modules enriched in
various signaling pathways, offering a sophisticated perspective on potential
therapeutic interventions.
While our computational approach provides valuable insights, it is essential
to acknowledge its inherent limitations. The in-silico methods necessitate
subsequent experimental validation, and the complexity of neurodegenerative
diseases demands multifaceted research approaches. The individual variability
in disease progression represents another critical factor that current computational
models cannot fully capture. MD simulations were not included in this study,
but they could provide deeper insights into ligand-protein dynamics, binding
stability, and long-term molecular interactions. Future research should prioritize
in vitro and in vivo studies to validate our computational predictions, particularly
focusing on the pharmacokinetics, neuroprotective effects, and blood-brain barrier
permeability of CR compounds. Investigating multi-target drug development strategies
may also enhance therapeutic efficacy, providing a foundation for clinical advancements
in LBD treatment.
Conclusions
In conclusion, our computational study provides groundbreaking insights into the potential therapeutic mechanisms of Coptidis Rhizoma (CR) compounds for Lewy body dementia (LBD) through advanced bioinformatics methodologies. By identifying 214 common targets between CR compounds and LBD, we unveiled a complex molecular network underlying neurodegeneration, with protein-protein interaction analysis highlighting critical hub genes including TARDBP, MAPT, GBA, SNCA, and PRKN that are pivotal in understanding the disease's molecular pathogenesis. Molecular docking simulations demonstrated remarkable binding interactions, particularly for compounds like Coptisine and Obacunone, which showed promising affinities for key proteins. Gene ontology and pathway enrichment analyses uncovered essential biological processes including neuronal apoptosis regulation, oxidative stress response, and synaptic transmission, offering potential therapeutic intervention targets. By bridging traditional herbal medicine with computational techniques, our research presents a novel approach to exploring LBD treatment strategies, integrating network pharmacology and molecular docking into a promising research framework. While our computational approach provides valuable insights, the need for experimental validation remains critical, suggesting future studies should focus on in vitro and in vivo investigations to confirm our computational predictions and explore the therapeutic potential of Coptidis Rhizoma compounds in addressing this complex neurodegenerative disorder.
Abbreviations
GBA – Glucocerebrosidase (also known as β-Glucosidase)
MAPT – Microtubule-Associated Protein Tau
TARDBP – TAR DNA-Binding Protein 43 (TDP-43)
SNCA – Alpha-Synuclein
PRKN – Parkin RBR E3 Ubiquitin Ligase
LBD: Lewy body dementia
CR: Coptidis Rhizoma
Declarations
Author Contributions
Conceptualization: P.S.; methodology: P.S.; software: P.S., A.N.; validation: P.S. and A.N.; formal analysis: P.S. and A.N.; investigation: P.S.; resources: A.N. and P.S.; data curation: P.S.; writing—original draft: A.N., P.S.; writing—review and editing: P.S.; visualization, P.S., A.A.; Project administration: P.S.; All authors have read and agreed to the published version of the manuscript.
Availability of Data and Materials
Not applicable.
Financial Support and Sponsorship
None.
Conflicts of Interest
All the authors declared that there are no conflicts of interest.
Ethical Approval and Informed Consent
Not applicable.
Consent for Publication
Not applicable.
References
1. Prasad S, Katta M, Abhishek S, Sridhar R, Valisekka S, Hameed M, et al. Recent advances in Lewy body dementia: a comprehensive review. Dis Mon, 2023, 69(5): 101441. [Crossref]
2. Lewy body dementia-Symptoms and causes-Mayo Clinic n.d. from [https://www.mayoclinic.org/diseases-conditions/lewy-body-dementia/symptoms-causes/syc-20352025.]
3. Lewy body dementia. National Institute of neurological disorders and stroke. from [https://www.ninds.nih.gov/health-information/disorders/lewy-body-dementia]
4. Treatment options–Lewy body dementia association. from [https://www.lbda.org/treatment-options]
5. Lewy body dementia-siagnosis and treatment-Mayo Clinic n.d. from [https://www.mayoclinic.org/diseases-conditions/lewy-body-dementia/diagnosis-treatment/drc-20352030]
6. Lewy body dementia: causes, symptoms, and diagnosis. National institute on aging n.D.
7. Simon C, Soga T, Okano H, & Parhar I. α-Synuclein-mediated neurodegeneration in dementia with Lewy bodies: the pathobiology of a paradox. Cell Biosci, 2021, 11(1): 196-208. [Crossref]
8. Harvey J, Pishva E, Chouliaras L, & Lunnon K. Elucidating distinct molecular signatures of Lewy body dementias. Neurobiol Dis, 2023, 188: 106337. [Crossref]
9. Meeus B, Theuns J, & Van Broeckhoven C. The genetics of dementia with Lewy bodies: what are we missing? Arch Neurol, 2012, 69(9): 1113-1118. [Crossref]
10. Sun Z, Yang H, & Chen S. Traditional Chinese medicine: a promising candidate for the treatment of Alzheimer's disease. Transl Neurodegener, 2013, 2(1): 6-16. [Crossref]
11. Bhushan B, Singh N, & Singh R. Traditional Chinese medicine: its growing potential in treating neurological disorders. Pharmacological Research - Modern Chinese Medicine, 2024, 11: 100422. [Crossref]
12. Wang J, Wang L, Lou G, Zeng H, Hu J, Huang Q, et al. Coptidis Rhizoma: a comprehensive review of its traditional uses, botany, phytochemistry, pharmacology and toxicology. Pharm Biol, 2019, 57(1): 193-225. [Crossref]
13. Singh K, Gupta J, Jain D, Chaudhary A, Kumar S, Sharma M, et al. Unveiling the therapeutic potential of traditional Chinese medicine Rhizome Coptidis: a comprehensive review. Pharmacological Research - Modern Chinese Medicine, 2024, 13: 100522. [Crossref]
14. Meng F, Wu Z, Yin Z, Lin L, Wang R, & Zhang Q. Coptidis Rhizoma and its main bioactive components: recent advances in chemical investigation, quality evaluation and pharmacological activity. Chinese Medicine, 2018, 13(1): 13-23. [Crossref]
15. Shahzadi Z, Yousaf Z, Anjum I, Bilal M, Yasin H, Aftab A, et al. Network pharmacology and molecular docking: combined computational approaches to explore the antihypertensive potential of Fabaceae species. Bioresour Bioprocess, 2024, 11(1): 53-64. [Crossref]
16. Long S, Ji S, Xue P, Xie H, Ma Y, & Zhu S. Network pharmacology and molecular docking analysis reveal insights into the molecular mechanism of shiliao decoction in the treatment of cancer-associated malnutrition. Frontiers in Nutrition, 2022, Volume 9. [Crossref]
17. Hamosh A, Scott A, Amberger J, Bocchini C, & McKusick V. Online mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res, 2005, 33: D514-517. [Crossref]
18. Darif D, Hammi I, Kihel A, El Idrissi Saik I, Guessous F, & Akarid K. The pro-inflammatory cytokines in COVID-19 pathogenesis: what goes wrong? Microb Pathog, 2021, 153: 104799. [Crossref]
19. The universal protein resource (UniProt). Nucleic Acids Res, 2008, 36: D190-195. [Crossref]
20. Ru J, Li P, Wang J, Zhou W, Li B, Huang C, et al. TCMSP: a database of systems pharmacology for drug discovery from herbal medicines. J Cheminform, 2014, 6: 13-23. [Crossref]
21. Daina A, Michielin O, & Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, 2017, 7(1): 42717. [Crossref]
22. Schwede T, Kopp J, Guex N, & Peitsch M. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res, 2003, 31(13): 3381-3385. [Crossref]
23. Davis A, Grondin C, Johnson R, Sciaky D, Wiegers J, Wiegers T, et al. Comparative toxicogenomics database (CTD): update 2021. Nucleic Acids Res, 2021, 49(D1): D1138-d1143. [Crossref]
24. The universal protein resource (UniProt) 2009. Nucleic Acids Res, 2009, 37: D169-174. [Crossref]
25. Oliveros, J. Venny. An interactive tool for comparing lists with venn’s diagrams. Scientific research publishing n.d. from [https://www.scirp.org/reference/referencespapers?referenceid=2904043]
26. Szklarczyk D, Kirsch R, Koutrouli M, Nastou K, Mehryary F, Hachilif R, et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res, 2023, 51(D1): D638-d646. [Crossref]
27. Chin C, Chen S, Wu H, Ho C, Ko M, & Lin C. CytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol, 2014, 8 Suppl 4: S11. [Crossref]
28. Hou J (2017). New approaches of protein function prediction from protein interaction networks. Chapter 1 - introduction. J. Hou, Academic Press: 1-20.
29. Sherman B, Hao M, Qiu J, Jiao X, Baseler M, Lane H, et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res, 2022, 50(W1): W216-w221. [Crossref]
30. Bader G, & Hogue C. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics, 2003, 4(1): 2-10. [Crossref]
31. Li T, Wernersson R, Hansen R, Horn H, Mercer J, Slodkowicz G, et al. A scored human protein–protein interaction network to catalyze genomic interpretation. Nature Methods, 2017, 14(1): 61-64. [Crossref]
32. Szklarczyk D, Gable A, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res, 2019, 47(D1): D607-d613. [Crossref]
33. Pettersen E, Goddard T, Huang C, Meng E, Couch G, Croll T, et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Science, 2021, 30(1): 70-82. [Crossref]
34. Goddard T, Huang C, Meng E, Pettersen E, Couch G, Morris J, et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Science, 2018, 27(1): 14-25. [Crossref]
35. Dev Sharma P, Alhudhaibi A, Al Noman A, Abdallah E, Taha T, & Sharma H. Systems biology-driven discovery of host-targeted therapeutics for oropouche virus: integrating network pharmacology, molecular docking, and drug repurposing. Pharmaceuticals, 2025, 18(5): 613-623.
36. O'Boyle N, Morley C, & Hutchison G. Pybel: a python wrapper for the OpenBabel cheminformatics toolkit. Chemistry Central Journal, 2008, 2(1): 5-16. [Crossref]
37. Vázquez-Jiménez L, Juárez-Saldivar A, Gómez-Escobedo R, Delgado-Maldonado T, Méndez-Álvarez D, Palos I, et al. Ligand-based virtual screening and molecular docking of benzimidazoles as potential inhibitors of triosephosphate isomerase identified new trypanocidal agents. Int J Mol Sci, 2022, 23(17): 10047. [Crossref]
38. Noman A, Sharma P, Mim T, Azad M, & Sharma H. Molecular docking and ADMET analysis of coenzyme Q10 as a potential therapeutic agent for Alzheimer’s disease. Aging Pathobiol Ther, 2024, 6(4): 170-182. [Crossref]
39. Srinivasan S, Sadasivam S, Gunalan S, Shanmugam G, & Kothandan G. Application of docking and active site analysis for enzyme linked biodegradation of textile dyes. Environ Pollut, 2019, 248: 599-608. [Crossref]
40. Naveed M, Din M, Aziz T, Javed T, Khan S, Naveed R, et al. Comparative analysis among the degradation potential of enzymes obtained from Escherichia coli against the toxicity of sulfur dyes through molecular docking. Zeitschrift für Naturforschung C, 2024, 79(7-8): 221-234. [Crossref]
41. Wang S, Jiang J, Li R, & Deng P. Docking-based virtual screening of TβR1 inhibitors: evaluation of pose prediction and scoring functions. BMC Chemistry, 2020, 14(1): 52-62. [Crossref]
42. Alhawarri M, Dianita R, Rawa M, Nogawa T, & Wahab H. Potential anti-cholinesterase activity of bioactive compounds extracted from Cassia grandis L.f. and Cassia timoriensis DC. Plants, 2023, 12(2): 344-354. [Crossref]
43. Kim T, Zhen J, Lee J, Bauer R, Lee C, Kwon B, et al. Influence of ligand's directional configuration, chrysenes as model compounds, on the binding activity with aryl hydrocarbon receptor. Sci Rep, 2020, 10(1): 13821. [Crossref]
44. Spassov D. Binding affinity determination in drug design: Insights from lock and key, induced fit, conformational selection, and inhibitor trapping models. Int J Mol Sci, 2024, 25(13): 7124-7135. [Crossref]
45. Grünberg R, Nilges M, & Leckner J. Flexibility and conformational entropy in protein-protein binding. Structure, 2006, 14(4): 683-693. [Crossref]
46. Benet L, Hosey C, Ursu O, & Oprea T. BDDCS, the rule of 5 and drugability. Adv Drug Deliv Rev, 2016, 101: 89-98. [Crossref]
47. Sumalapao D, Villarante N, Agapito J, Asaad A, & Gloriani N. Topological polar surface area, molecular weight, and rotatable bond count account for the variations in the inhibitory potency of antimycotics against Microsporum canis. J. Pure Appl. Microbiol, 2020, 14(1): 247-254. [Crossref]
48. Prasanna S, & Doerksen R. Topological polar surface area: a useful descriptor in 2D-QSAR. Curr Med Chem, 2009, 16(1): 21-41. [Crossref]
49. Xu X, Zhang W, Huang C, Li Y, Yu H, Wang Y, et al. A novel chemometric method for the prediction of human oral bioavailability. Int J Mol Sci, 2012, 13(6): 6964-6982. [Crossref]
50. Jia C, Li J, Hao G, & Yang G. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov Today, 2020, 25(1): 248-258. [Crossref]
51. Mittal A, Ghai R, Srivastava A, Ghosh D, & Nagarajan K (2023). Pharmacokinetics and pharmacodynamics: fundamentals and role(s) in drug discovery and development. Recent Advances in Pharmaceutical Innovation and Research. Springer Nature Singapore: 357-393.
52. Pantsar T, & Poso A. Binding affinity via docking: Fact and fiction. Molecules, 2018, 23(8): 1899-1905. [Crossref]
53. Pedicone C, Weitzman S, Renton A, & Goate A. Unraveling the complex role of MAPT-containing H1 and H2 haplotypes in neurodegenerative diseases. Mol Neurodegener, 2024, 19(1): 43-55. [Crossref]
54. Strang K, Golde T, & Giasson B. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab Invest, 2019, 99(7): 912-928. [Crossref]
55. Walton R, Koga S, Beasley A, White L, Griesacker T, Murray M, et al. Role of GBA variants in Lewy body disease neuropathology. Acta Neuropathologica, 2024, 147(1): 54-60. [Crossref]
56. Zheng W, & Fan D. Glucocerebrosidase mutations cause mitochondrial and lysosomal dysfunction in Parkinson's disease: Pathogenesis and therapeutic implications. Front Aging Neurosci, 2022, 14: 851135. [Crossref]
57. Johnson P, Weinreb N, Cloyd J, Tuite P, & Kartha R. GBA1 mutations: Prospects for exosomal biomarkers in α-synuclein pathologies. Mol Genet Metab, 2020, 129(2): 35-46. [Crossref]
58. Cabeza-Arvelaiz Y, Fleming S, Richter F, Masliah E, Chesselet M, & Schiestl R. Analysis of striatal transcriptome in mice overexpressing human wild-type alpha-synuclein supports synaptic dysfunction and suggests mechanisms of neuroprotection for striatal neurons. Mol Neurodegener, 2011, 6: 83-94. [Crossref]
59. Brás J, Gibbons E, & Guerreiro R. Genetics of synucleins in neurodegenerative diseases. Acta Neuropathol, 2021, 141(4): 471-490. [Crossref]
60. Terešak P, Lapao A, Subic N, Boya P, Elazar Z, & Simonsen A. Regulation of PRKN-independent mitophagy. Autophagy, 2022, 18(1): 24-39. [Crossref]
61. Quinn P, Moreira P, Ambrósio A, & Alves C. 61PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol Commun, 2020, 8(1): 189-193. [Crossref]
62. Ma K, Chen G, Li W, Kepp O, Zhu Y, & Chen Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front Cell Dev Biol, 2020, 8: 467-478. [Crossref]
63. Jo M, Lee S, Jeon Y, Kim S, Kwon Y, & Kim H. The role of TDP-43 propagation in neurodegenerative diseases: integrating insights from clinical and experimental studies. Experimental & Molecular Medicine, 2020, 52(10): 1652-1662. [Crossref]
64. Prasad A, Bharathi V, Sivalingam V, Girdhar A, & Patel B. Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front Mol Neurosci, 2019, 12: 25-34. [Crossref]
65. Suk T, & Rousseaux M. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol Neurodegener, 2020, 15(1): 45-56. [Crossref]
66. Xiao N, & Le Q. Neurotrophic factors and their potential applications in tissue regeneration. Arch Immunol Ther Exp (Warsz), 2016, 64(2): 89-99. [Crossref]
67. Bathina S, & Das U. Brain-derived neurotrophic factor and its clinical implications. Arch Med Sci, 2015, 11(6): 1164-1178. [Crossref]
68. Olufunmilayo E, Gerke-Duncan M, & Holsinger R. Oxidative stress and antioxidants in neurodegenerative disorders. Antioxidants (Basel), 2023, 12(2): 517-523. [Crossref]
69. Dash U, Bhol N, Swain S, Samal R, Nayak P, Raina V, et al. Oxidative stress and inflammation in the pathogenesis of neurological disorders: mechanisms and implications. Acta Pharm Sin B, 2025, 15(1): 15-34. [Crossref]
70. Yan Z, & Rein B. Mechanisms of synaptic transmission dysregulation in the prefrontal cortex: pathophysiological implications. Mol Psychiatry, 2022, 27(1): 445-465. [Crossref]
71. Lepeta K, Lourenco M, Schweitzer B, Martino Adami P, Banerjee P, Catuara-Solarz S, et al. Synaptopathies: synaptic dysfunction in neurological disorders – a review from students to students. Journal of Neurochemistry, 2016, 138(6): 785-805. [Crossref]
72. BIOVIA Discovery studio. Dassault systèmes n.d. from [Crossref]
73. Kaba B, Pinet N, Lelandais G, Sigayret A, & Berry A. Clustering gene expression data using graph separators. In Silico Biol, 2007, 7(4-5): 433-452.
74. Wu G, Robertson D, Brooks C 3rd, & Vieth M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-a CHARMm-based MD docking algorithm. J Comput Chem, 2003, 24(13): 1549-1562. [Crossref]
75. Brooks B, Brooks C 3rd, Mackerell A Jr., Nilsson L, Petrella R, Roux B, et al. CHARMM: the biomolecular simulation program. J Comput Chem, 2009, 30(10): 1545-1614. [Crossref]
76. Yang J, Roy A, & Zhang Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics, 2013, 29(20): 2588-2595. [Crossref]
77. Laskowski R, Jabłońska J, Pravda L, Vařeková R, & Thornton J. PDBsum: structural summaries of PDB entries. Protein Sci, 2018, 27(1): 129-134. [Crossref]