Open Access | Review
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
Proteostasis in aging: mechanistic insights and therapeutic opportunities
* Corresponding author: Gaurav N. Kasar
Mailing address: Department of Pharmacology, Divine College of Pharmacy, Satana (Nashik), India.
Email: gauravkasar008@gmail.com
Received: 17 December 2024 / Revised: 06 January 2024 / Accepted: 10 January 2025 / Published: 28 March 2025
DOI: 10.31491/APT.2025.03.165
Abstract
Proteostasis, the dynamic balance of protein synthesis, folding, and degradation, is fundamental to cellular homeostasis and organismal health. Aging disrupts proteostasis networks, leading to the accumulation of misfolded and aggregated proteins, which plays a central role in age-related dysfunction and the onset of diseases such as neurodegenerative and metabolic disorders. This review comprehensively explores the components and regulatory mechanisms of proteostasis networks, including key proteolytic systems like the ubiquitin-proteasome system (UPS) and autophagy, as well as the role of molecular chaperones in maintaining protein folding. We discuss hallmark features of aging-related proteostasis dysfunction and highlight its implications in major age-associated diseases, particularly neurodegenerative conditions like Alzheimer’s and Parkinson’s, and metabolic disorders such as diabetes and obesity. Additionally, emerging therapeutic strategies aimed at restoring proteostasis for healthy aging are examined, focusing on targeting chaperones, enhancing proteolytic systems, and modulating protein folding pathways. Advances in transcription factor regulation, proteasome activators, and autophagy modulators, as well as promising approaches involving small molecules and gene therapy, are discussed. Finally, we outline future directions and conclude that targeting proteostasis represents a promising avenue for improving health span and mitigating age-related diseases.
Keywords
Proteostasis, proteolytic, dysfunction, neurodegenerative, age-related diseases
Introduction
Proteostasis, or protein homeostasis,
is a critical process involving the synthesis,
folding, trafficking, and degradation of proteins
to maintain a functional proteome. It plays an
essential role in cellular health by ensuring
proteins achieve and retain their proper
three-dimensional structure, which is necessary
for their biological function [1]. The proteostasis
network includes molecular chaperones that assist
in protein folding, the ubiquitin-proteasome
system for protein degradation, and pathways
that address protein misfolding and aggregation.
These systems work in concert to manage protein
quality and prevent the accumulation of defective
proteins, which can disrupt cellular functions
and lead to diseases, particularly neurodegenerative
conditions like Alzheimer’s and Parkinson’s [2, 3].
The efficiency of the proteostasis machinery is
challenged by factors such as aging, environmental
stress, and disease. As organisms age, the decline
in proteostasis capacity contributes to the accumulation
of misfolded or aggregated proteins, which are hallmarks
of several age-related disorders. For example, molecular
chaperones and proteasomal activity decline with age,
making cells less capable of managing protein quality.
Understanding the mechanisms of proteostasis and its
regulation not only provides insights into fundamental
biology but also has therapeutic implications for
improving health and treating diseases associated
with proteome instability [4-6].
As organisms age, the mechanisms
that regulate proteostasis protein
synthesis, folding, and degradation
become less effective, leading to an
accumulation of misfolded and damaged
proteins. This imbalance, often termed
"proteostasis collapse", significantly
impacts cellular function, longevity,
and susceptibility to diseases such as
neurodegeneration, cancer, and cardiovascular
disorders. Proteostasis is maintained by an
intricate network of molecular chaperones,
the ubiquitin-proteasome system (UPS), and
autophagy pathways. With aging, these systems
face increased oxidative stress, damage from
reactive oxygen species (ROS), and a decline
in efficiency [7]. The UPS, critical for degrading
short-lived or misfolded proteins, becomes impaired,
leading to an accumulation of ubiquitinated proteins
and aggregates that disrupt cellular homeostasis.
Similarly, autophagy, which removes large protein
aggregates and damaged organelles, declines with age,
further exacerbating proteotoxic stress. These deficiencies
contribute to the hallmark signs of aging, such as cellular
senescence, chronic inflammation, and tissue dysfunction.
The decline in proteostasis significantly contributes to
cellular aging and chronic diseases. For instance, in
neurodegenerative diseases like Alzheimer’s and Parkinson’s,
protein aggregates such as amyloid-beta and alpha-synuclein
disrupt cellular function. In cancer, although proteostasis
mechanisms are hyper activated to support the rapid proliferation
of cells, aging reduces their efficacy, potentially leading to
tumorigenesis. Moreover, systemic inflammation associated with
aging, termed "inflammation," is partially driven by the phototoxic
stress caused by impaired protein homeostasis. Interventions targeting
these pathways, such as enhancing autophagy or proteasomal activity,
have shown potential in extending lifespan and improving health span
in model organisms [8, 9].
The primary objective of this review is to explore
the intricate relationship between proteostasis
and aging, emphasizing how the regulation of protein
homeostasis impacts cellular health and overall longevity.
It aims to examine the role of proteostasis in maintaining
protein quality and stability and its deterioration
as a hallmark of aging. Furthermore, the review
investigates the implications of disrupted
proteostasis for human health, with a focus
on age-related diseases such as neurodegeneration,
cancer, and cardiovascular disorders. By understanding
the mechanisms underlying proteostasis collapse and its
association with aging, the review seeks to highlight
potential therapeutic strategies to enhance proteostasis,
promote healthy aging, and mitigate the progression of
age-related diseases.
Proteostasis networks: components and mechanisms
Molecular chaperones play an essential role in protein folding and quality control by assisting in the proper folding of newly synthesized proteins, ensuring their stability, and preventing the aggregation of misfolded proteins [3]. These chaperones act in several ways, such as by binding to nascent proteins and preventing their premature folding or degradation, or by facilitating the refolding of misfolded proteins under stress conditions like heat or oxidative stress. They help maintain proteostasis, which is crucial for cellular function, particularly in cells subjected to environmental stressors, such as neurons. Heat shock proteins (HSPs) are among the most well-known molecular chaperones, and they are named for their increased expression in response to heat stress. HSPs, including Hsp70, Hsp90, and small HSPs, provide a protective environment that allows proteins to adopt their functional conformations. When refolding is impossible, chaperones direct misfolded proteins to degradation pathways, such as the ubiquitin-proteasome system (UPS) or autophagy, preventing the accumulation of toxic aggregates that can impair cellular function [10, 11] . Chaperones are involved in a range of protein quality control mechanisms, such as disaggregating protein clusters or guiding defective proteins to degradation pathways. For example, misfolded proteins are often marked by ubiquitination, which signals the proteasome to degrade them, while other misfolded proteins are transported to lysosomes for breakdown through autophagy. This intricate system is particularly vital for preventing neurodegenerative diseases associated with protein misfolding, such as Alzheimer’s and Parkinson’s diseases [12] (Table 1).
Table 1.
Various examples of Chaperone involved in protein folding and quality control.
| Chaperone | Role | Function/Mechanism | References |
|---|---|---|---|
| HSP70 | Protein folding, refolding, stabilization | HSP70 binds to nascent polypeptides, preventing premature folding and aggregation, and refolds misfolded proteins. | [3, 10] |
| HSP90 | Protein maturation, stability | HSP90 assists in the maturation of client proteins, including kinases, and stabilizes misfolded proteins. | [11, 12] |
| sHSPs (small heat shock proteins) | Prevent aggregation, assist in protein folding | sHSPs prevent aggregation by binding to unfolded proteins under stress and facilitate their refolding. | [10, 12] |
| HSP60 (GroEL) | Protein folding and assembly | HSP60 acts as a molecular chaperonin, forming a chamber where proteins are enclosed and properly folded. | [13, 14] |
| HSP100 | Protein disaggregation, proteostasis | HSP100 proteins assist in the disaggregation of protein complexes and refold proteins under stress conditions. | [15] |
| TriC/CCT | Protein folding, oligomeric assembly | TriC/CCT is a chaperonin complex that assists in the folding of actin, tubulin, and other cytoskeletal proteins. | [16, 17] |
| HSP40 | Co-chaperone, assists HSP70 | HSP40 interacts with HSP70, guiding substrate proteins to HSP70 for folding and preventing aggregation. | [18, 19] |
Proteolytic systems
Ubiquitin-proteasome system (UPS)
The UPS is an essential intracellular proteolytic pathway that plays a central role in maintaining proteostasis the delicate balance of protein synthesis, folding, and degradation within cells. The UPS ensures the selective degradation of damaged, misfolded, or unnecessary proteins, preventing the accumulation of toxic aggregates that can disrupt cellular function. The system operates through a tightly regulated sequence of events. It begins with the activation of ubiquitin, a small regulatory protein, by the E1 ubiquitin-activating enzyme in an ATP-dependent manner. This activated ubiquitin is transferred to the E2 ubiquitin-conjugating enzyme, and finally, the E3 ubiquitin ligase facilitates the attachment of ubiquitin to specific substrate proteins, determining their fate (Figure 1). Proteins tagged with polyubiquitin chains are subsequently recognized and degraded by the 26S proteasome, a highly specialized protease complex. The degradation process releases peptides, which are further broken down into amino acids for recycling.
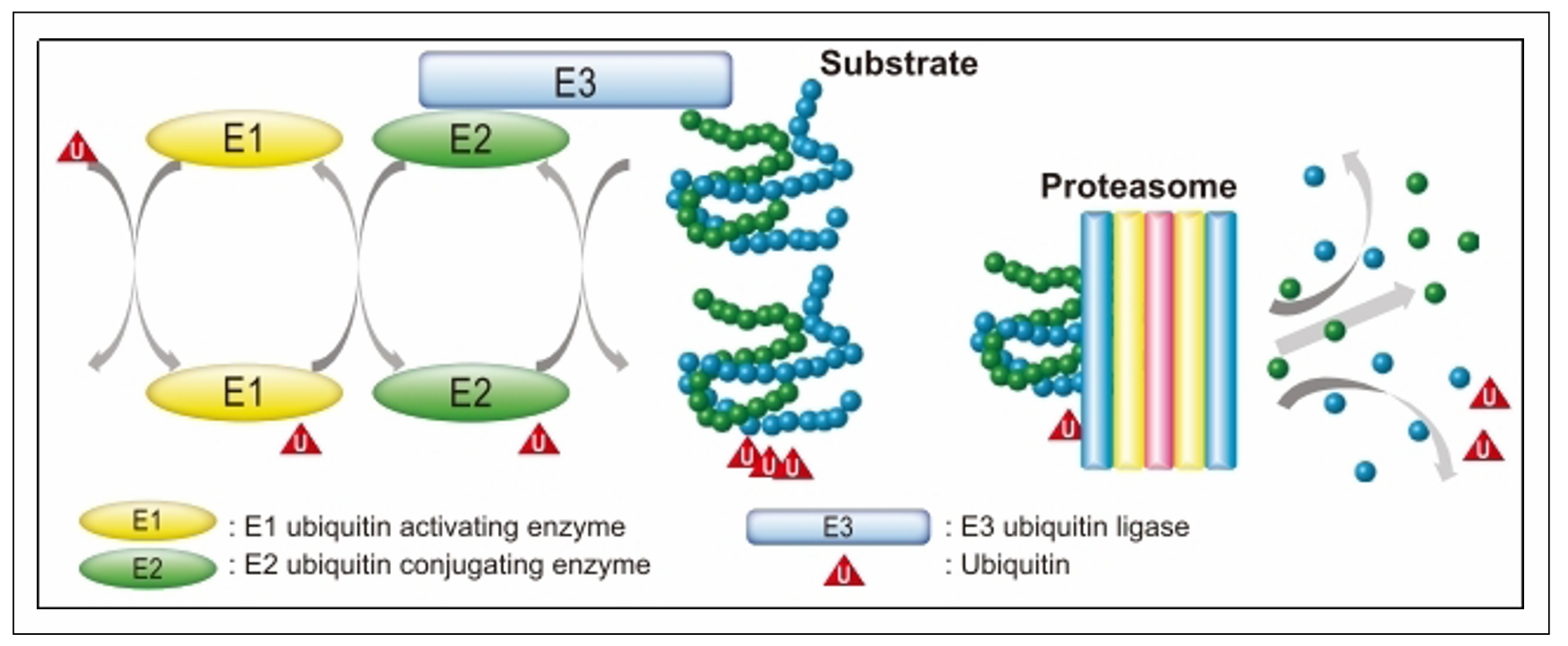
Figure 1. Ubiquitin-proteasome system (UPS).
E1 (ubiquitin-activating enzyme): E1
is the initial enzyme in the ubiquitination cascade.
It activates ubiquitin in an ATP-dependent manner,
forming a high-energy thioester bond between the
C-terminal glycine residue of ubiquitin and a cysteine
residue on the E1 enzyme. This step primes ubiquitin
for subsequent transfer and is critical for initiating
the ubiquitination process. E1 exists in limited numbers
in cells, as a single E1 enzyme can activate multiple
ubiquitin molecules for further transfer to E2 enzymes.
The activation involves two steps: Adenylation of the
ubiquitin molecule’s C-terminal glycine using ATP, producing
ubiquitin-AMP and Formation of the thioester bond between
ubiquitin and the active site cysteine of E1, releasing AMP [20].
E2 (ubiquitin-conjugating enzyme): The E2 enzyme
receives ubiquitin from E1 through a transthiolation
reaction, where the activated ubiquitin is transferred
to an active site cysteine on E2. E2 enzymes are responsible
for carrying ubiquitin to E3 ligases and determining the
type of ubiquitin chain linkage that will be added to the
substrate. E2 enzymes also dictate the topology of the
ubiquitin chain, influencing the fate of the ubiquitinated
protein. There are multiple E2 enzymes in cells, each
specialized for different functions, such as monoubiquitination,
polyubiquitination, or specific chain formations like K48-linked
chains (for degradation) or K63-linked chains (for signaling) [21, 22].
E3 (ubiquitin ligase): E3 ligases are responsible
for the substrate specificity of ubiquitination.
These enzymes recognize target proteins through
specific degradation signals or motifs, such as
phosphorylation tags or hydrophobic patches
exposed due to protein misfolding. E3 ligases
catalyze the transfer of ubiquitin from the E2
enzyme to the target protein, either directly
or indirectly. Types of E3 ligases: HECT (homologous
to E6-AP carboxyl terminus): These E3 ligases form
a thioester intermediate with ubiquitin before
transferring it to the substrate. RING (really
interesting new gene) and RBR (RING-between-RING):
These E3s facilitate the direct transfer of ubiquitin
from E2 to the substrate without forming a thioester
bond. The substrate recognition of E3 ligases is
critical for cellular regulation, as it ensures that
only proteins marked for degradation are ubiquitinated.
Examples include the MDM2 ligase, which targets p53,
and Parkin, associated with mitochondrial quality control.
The coordinated action of E1, E2, and E3 enzymes ensures
the selectivity and efficiency of the UPS. This specificity
is vital for maintaining cellular homeostasis, regulating
the cell cycle, controlling apoptosis, responding to stress,
and modulating signal transduction. Dysregulation in any
step of the ubiquitination process can lead to diseases
such as cancer, neurodegenerative disorders (e.g., Parkinson’s
disease due to Parkin dysfunction), and immune deficiencies [23, 24].
The UPS is fundamental to numerous biological
processes, including cell cycle regulation,
DNA repair, apoptosis, immune responses, and
adaptation to stress. By removing oxidative
damaged or misfolded proteins, the system
protects cells from proteotoxicity, which
is particularly critical in post-mitotic
cells such as neurons. However, during
aging, the efficiency of the UPS declines
due to decreased expression of proteasomal
subunits, reduced activity of E3 ligases,
and accumulation of inhibitory substrates.
This decline leads to the buildup of misfolded
and aggregated proteins, contributing to cellular
dysfunction and aging-related diseases, particularly
neurodegenerative disorders. For instance, in
Alzheimer’s disease, the UPS struggles to manage
the accumulation of amyloid-beta and tau proteins,
while in Parkinson’s disease; alpha-synuclein
aggregates overwhelm the system. Similarly,
Huntington’s disease features polyglutamine
expansions that resist proteasomal degradation,
exacerbating the proteostasis imbalance [25, 26].
Research has also highlighted the
interplay between the UPS and other
proteolytic pathways, such as the
autophagy-lysosome system, in managing
cellular protein turnover. Together,
these systems form a dynamic network to
maintain proteostasis. However, with aging,
this synergy is impaired, further exacerbating
proteotoxic stress. Efforts to counteract this
age-associated decline have inspired therapeutic
approaches targeting the UPS. For example, small
molecules that activate the proteasome or enhance
the activity of E3 ligases show promise in clearing
pathological proteins and restoring proteostasis [27].
Additionally, compounds that enhance the cross-talk between
UPS and autophagy could provide dual benefits in
promoting protein clearance. While proteasome inhibitors
like bortezomib are effective in cancer therapies
by inducing stress in rapidly dividing cells, their
application in aging is limited due to the potential
for exacerbating proteotoxic stress. Emerging research
emphasizes the importance of understanding the molecular
and structural intricacies of the UPS for developing
precision therapies. Future strategies could include
personalized approaches that target specific components
of the UPS to enhance its activity in aging tissues or
mitigate its dysfunction in disease states. The UPS
remains a critical area of study for interventions
aimed at extending health span and combating age-related pathologies [28, 29].
Autophagy-lysosome pathway (ALP)
The ALP is a fundamental cellular
process that maintains homeostasis
by degrading and recycling damaged
proteins, dysfunctional organelles,
and other cellular debris. This pathway
operates by forming double-membraned
vesicles, known as auto phagosomes,
which engulf the targeted materials.
Auto phagosomes subsequently fuse with
lysosomes, specialized organelles containing
hydrolytic enzymes, to form autolysosomes.
Inside these autolysosomes, the contents
are degraded into basic building blocks
like amino acids and lipids, which are
then recycled to support cellular functions
and energy metabolism. This pathway is
essential for maintaining proteostasis,
adapting to stress, and regulating metabolism [30-32].
Autophagy is categorized into three types:
macroautophagy, which targets large aggregates
and organelles; microautophagy, where lysosomes
directly engulf cytoplasmic material; and
chaperone-mediated autophagy (CMA), a selective
process where specific proteins are delivered to
lysosomes by chaperones like Hsc70 and processed
via LAMP-2A receptors. The ALP is tightly regulated
by pathways such as mTOR, which inhibits autophagy
under nutrient-rich conditions, and AMPK, which
activates it under energy stress. Beclin-1 is a
critical initiator of autophagosome formation [33].
Autophagy is a critical cellular process
that relies on various proteolytic enzymes
to maintain cellular homeostasis and function.
These enzymes play essential roles in degrading
damaged proteins and organelles, thereby supporting
proteostasis and cellular health. As individuals age,
the activity of these proteolytic enzyme declines,
contributing to the accumulation of damaged proteins
and dysfunctional cellular components. This decline
in proteolytic activity is a key factor in the aging
process and is linked to various age-related diseases.
Table 2 provides examples of proteolytic enzymes involved
in aging, along with their specific roles in maintaining
cellular function. Autophagy efficiency declines with age,
contributing to the accumulation of protein aggregates and
damaged organelles. This decline exacerbates cellular
dysfunction and is linked to age-related diseases. For
example, in neurodegenerative diseases like Alzheimer’s,
Parkinson’s, and Huntington’s, impaired autophagy leads
to the accumulation of toxic protein aggregates.
In metabolic disorders, autophagy dysfunction affects
insulin signaling and lipid metabolism, aggravating
conditions like diabetes and obesity. Interestingly,
in cancer, autophagy has a dual role: it can suppress
tumor initiation by clearing damaged components but may
promote survival in established tumors under metabolic
stress [34, 35]. Enhancing autophagy offers promising
therapeutic potential. Pharmacological agents like
rapamycin, which inhibits mTOR, and metformin, an AMPK
activator, have shown efficacy in boosting autophagy and
improving lifespan in preclinical models. Additionally,
CMA enhancement by increasing LAMP-2A expression holds
potential for selective degradation of toxic proteins.
However, balancing autophagy is crucial to avoid excessive
activation, which can lead to autophagic cell death.
Continued research into autophagy modulation is key
for developing therapies to combat aging-related diseases
and improve health span [36-38].
Table 2.
Examples of proteolytic enzymes involved in aging, along with their roles.
| Proteolytic enzyme | Function | Role in aging | References |
|---|---|---|---|
| Lon protease | Mitochondrial protease that degrades misfolded mitochondrial proteins | Decline in Lon protease activity with age contributes to mitochondrial dysfunction and oxidative stress, impacting cellular health | [39] |
| Proteasome | Multimeric enzyme complex that degrades damaged or unneeded proteins tagged by ubiquitin | Age-related decline in proteasomal activity leads to the accumulation of damaged proteins, contributing to age-related diseases like neurodegeneration | [40] |
| Aspartic proteases (e.g., Cathepsin D) | Acidic proteases involved in protein degradation within lysosomes | Reduced cathepsin D activity in aging affects protein turnover and contributes to the build-up of damaged proteins in neurons, contributing to neurodegenerative diseases | [41] |
| Metalloproteinases (e.g., MMP-9) | Enzyme responsible for breaking down extracellular matrix proteins | Overexpression of MMP-9 in aging promotes tissue degradation and has been implicated in diseases such as Alzheimer’s and cardiovascular aging | [42] |
| Deubiquitinases (e.g., USP14) | Proteases that remove ubiquitin from proteins, regulating protein degradation via the proteasome | Dysregulation of deubiquitinases like USP14 in aging can lead to the accumulation of damaged proteins and cellular dysfunction | [43] |
| Caspases | Cysteine-dependent proteases involved in apoptosis and cell death | Age-related activation of caspases contributes to neuronal loss and tissue degeneration seen in neurodegenerative diseases and other aging-associated pathologies | [44] |
| ClpXP protease | ATP-dependent protease that degrades abnormal proteins in bacteria and mitochondria | Decline in mitochondrial ClpXP activity during aging impairs protein quality control in mitochondria, contributing to mitochondrial dysfunction and cellular aging | [45] |
Regulatory mechanisms of proteostasis
Transcription factors: HSF-1, NRF2, and others
HSF-1 (Heat Shock Factor 1) is
a key transcription factor in
the heat shock response (HSR),
which is activated when cells
experience proteotoxic stress,
such as heat shock, oxidative damage,
or heavy metal exposure. Under normal
conditions, HSF-1 is kept inactive in
the cytoplasm. When stress occurs,
HSF-1 undergoes trimerization, a process
that leads to its activation and translocation
to the nucleus. Once in the nucleus, HSF-1
binds to heat shock elements (HSEs) in the
promoter regions of heat shock protein (HSP)
genes, initiating their transcription. These
HSPs, such as HSP70 and HSP90, are molecular
chaperones that help proteins fold correctly
and protect against aggregation. HSF-1 activation
has been linked to increased lifespan in organisms
like C. elegans and Drosophila by enhancing cellular
resistance to stress and reducing protein misfolding
and aggregation, which are hallmarks of aging and
neurodegenerative diseases. Furthermore, HSF-1 plays
a role in regulating the expression of other stress-related
genes, contributing to cellular maintenance during aging.
Factor erythroid 2-related factor 2 (NRF2) is a key regulator
of the antioxidant response, orchestrating the expression of
genes involved in oxidative stress defense. Under normal conditions,
NRF2 is kept inactive in the cytoplasm through binding to Keap1
(Kelch-like ECH-associated protein 1). Upon oxidative stress or
electrophilic stress, Keap1 undergoes conformational changes that
release NRF2, allowing it to translocate to the nucleus. In the
nucleus, NRF2 binds to antioxidant response elements
(AREs) in the promoter regions of genes encoding antioxidant
enzymes (such as superoxide dismutase and glutathione S-transferase).
NRF2 activation is a critical response to counteract oxidative damage
caused by reactive oxygen species (ROS). The decline in NRF2 function
during aging leads to an accumulation of oxidative damage, which accelerates
aging and is implicated in neurodegenerative diseases such as Parkinson’s
and Alzheimer’s disease. Boosting NRF2 activity has been suggested as a
therapeutic strategy to mitigate oxidative stress-related damage and enhance
longevity [46-49].
Other transcrcontribute to stress responses and
proteostasis regulation. For example, ATF4 (activating
transcription factor 4) plays a central role in the unfolded
protein response (UPR), a cellular mechanism activated under
conditions of endoplasmic reticulum (ER) stress when misfolded
proteins accumulate in the ER. ATF4 promotes the expression of
genes that facilitate protein folding, degradation, and export,
helping restore cellular homeostasis during stress. Additionally,
FOXO transcription factors are involved in longevity regulation
and the cellular response to stress. FOXO factors are activated
by oxidative stress and promote autophagy, apoptosis, and the
maintenance of cellular repair processes. Their role in aging
is crucial, as they modulate both protein quality control and
stress resilience (Figure 2) [50].
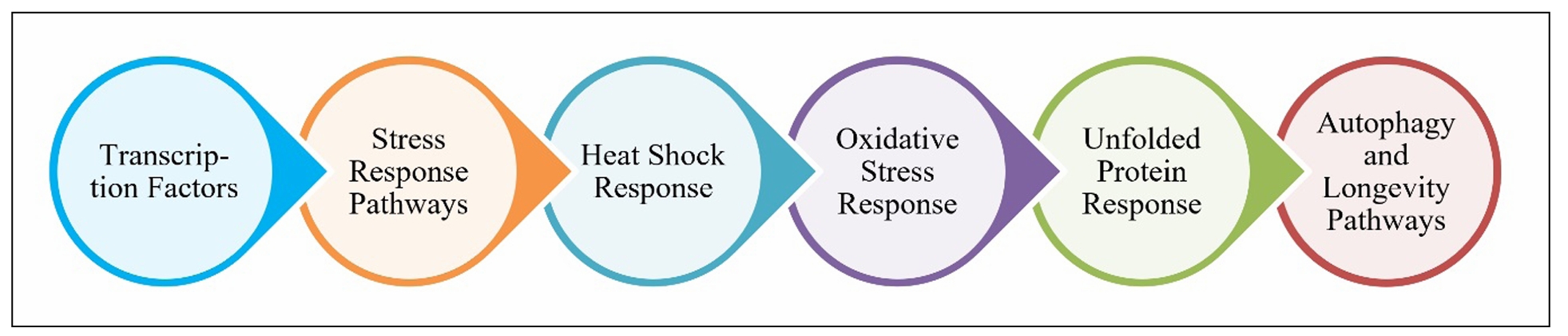
Figure 2. Regulatory mechanisms of proteostasis.
Stress response pathways
Stress response pathways are fundamental
to maintaining proteostasis, especially
under cellular stress conditions like
oxidative stress, heat shock, nutrient
deprivation, or the accumulation of damaged
proteins. These pathways are regulated by
transcription factors like HSF-1 and NRF2,
and their activation ensures cellular homeostasis.
Below are key stress response pathways:
Heat shock response (HSR): The heat shock
response, regulated by HSF-1, is one of the
first lines of defense against proteotoxic
stress, particularly when proteins begin to
misfolded under high temperatures or stress
conditions. HSF-1 activates the transcription of
heat shock proteins (HSPs), which act as molecular
chaperones to help in protein folding and prevent
the aggregation of misfolded proteins. In aging,
the efficiency of the heat shock response declines,
contributing to the accumulation of misfolded proteins
that exacerbate age-related diseases such as Alzheimer’s,
Huntington’s, and Parkinson’s diseases. Enhancing the heat
shock response by activating HSF-1 has been shown to extend
lifespan in model organisms and could be a therapeutic approach
for combating age-related diseases [46, 51, 52].
Oxidative stress response: The oxidativrimarily
regulated by NRF2, is activated in response to increased
levels of reactive oxygen species (ROS). ROS are
byproducts of normal cellular metabolism, but their
accumulation due to mitochondrial dysfunction,
environmental stress, or aging can cause cellular
damage. NRF2 regulates the transcription of
antioxidant genes that protect cells from oxidative
damage. The ability to maintain NRF2 activation
diminishes with age, contributing to oxidative damage
and inflammation, which accelerates aging and the
development of neurodegenerative diseases. NRF2
activation has thus become a promising target for
therapies aimed at increasing cellular resistance
to oxidative stress and promoting healthy aging [53, 54].
Unfolded protein response (UPR): The misfolded
or unfolded proteins accumulate in the endoplasmic
reticulum (ER). The UPR involves three key sensor
proteins: IRE1, PERK, and ATF6, which help manage
protein folding, quality control, and degradation.
While the UPR can be protective by restoring cellular
function and preventing damage, chronic or excessive
UPR activation leads to apoptosis and has been linked
to aging and diseases like Alzheimer’s and Parkinson’s.
The efficiency of the UPR declines with age, contributing
to protein aggregation and cellular dysfunction. Enhancing
UPR signaling through modulation of its key components
could hold therapeutic potential in aging-related diseases [55, 56].
Autophagy and longevity pathways: Auto degradation
process that helps clear damaged proteins, organelles,
and other cellular debris. Under stress, the autophagic
process is upregulated, helping to maintain cellular
homeostasis. The FOXO transcription factors play a
critical role in promoting autophagy under stress
conditions like oxidative damage or nutrient scarcity.
In aging, the efficiency of autophagy decreases,
leading to the accumulation of dysfunctional cellular
components. This decline in autophagy has been linked
to age-related diseases such as neurodegenerative
disorders, cardiovascular diseases, and cancer.
Strategies aimed at enhancing autophagic activity
have shown promise in extending lifespan and
mitigating the effects of aging. These transcription
factors and stress response pathways are central to
maintaining proteostasis, particularly under stress conditions [57].
Hallmarks of aging-related proteostasis dysfunction
Impaired protein folding
Proteins must fold into specific three-dimensional structures to perform their functions correctly. This folding process is highly dynamic and relies on molecular chaperones like HSP70, HSP90, and small heat shock proteins, which assist in maintaining the correct protein conformation under stressful conditions. With aging, the efficiency of these chaperones decreases, leading to an increased risk of proteins misfolding, which contributes significantly to aging-related diseases such as Alzheimer’s, Parkinson’s, and Huntington’s diseases. As aging progresses, the cellular machinery that governs protein folding, including molecular chaperones and the endoplasmic reticulum (ER) chaperone system, deteriorates. This deterioration impairs the cell’s ability to cope with misfolded proteins, leading to a decline in cellular function and an accumulation of proteotoxic species. These misfolded proteins can become toxic, disrupting cellular homeostasis and triggering pathways such as the unfolded protein response (UPR), which, when chronically activated, can lead to cellular apoptosis and tissue dysfunction [58, 59]. The unfolded protein response (UPR) is a critical cellular stress response triggered when the load of misfolded proteins overwhelms the protein folding machinery. In aging, the UPR becomes less effective, leading to a buildup of unfolded or misfolded proteins, which exacerbates the damage to cells and tissues. Chronic activation of the UPR is linked to various age-related diseases, including neurodegenerative disorders. The UPR and its relationship with aging highlights how impaired protein folding is a central feature of proteostasis dysfunction in aging [60, 61].
Accumulation of misfolded or aggregated proteins
The accumulation of misfolded and aggregated proteins is one of the most profound hallmarks of aging-related proteostasis dysfunction. Misfolded proteins, if not refolded correctly or degraded, tend to aggregate and form inclusion bodies, which are toxic to cells. These aggregates often resist degradation by the UPS or autophagy, which are the primary pathways responsible for the removal of damaged proteins. Over time, the accumulation of protein aggregates can overwhelm the cellular degradation systems, leading to cellular damage, inflammation, and organ dysfunction [46, 52]. A key example of this is amyloid plaques in Alzheimer’s disease, composed primarily of amyloid-β (Aβ), which are the result of the aggregation of misfolded Aβ peptides. In Parkinson’s disease, alpha-synuclein forms aggregates known as Lewy bodies. These aggregates interfere with normal cellular processes such as protein degradation and can lead to neurodegeneration and cell death. The accumulation of these protein aggregates is particularly problematic in neurons, which have limited regenerative capabilities. As cells age, their ability to clear protein aggregates diminishes, contributing to the onset of neurodegenerative diseases [62]. The role of impaired proteostasis in aging is especially evident in the context of the proteasome and autophagy, two key pathways that regulate the degradation of damaged proteins. With aging, the proteasome’s efficiency decreases, and autophagic flux slows down, both of which contribute to the accumulation of protein aggregates. This impairment is a central feature of age-related diseases such as neurodegeneration, cardiovascular disease, and even cancer. For instance, impaired protein quality control mechanisms like chaperone function and autophagy can lead to the accumulation of misfolded proteins, which is especially detrimental in neurons and muscles. The consequences of proteostasis imbalance are evident across systems such as the nervous, cardiovascular, and metabolic systems. Table 3 outlines the key systems affected by the decline in proteostasis and their associated impact on cellular functions.
Table 3.
Systems affected by proteostasis decline.
| System affected | Effect of proteostasis decline | Mechanisms involved | Disease implications | References |
|---|---|---|---|---|
| Neurons | Accumulation of misfolded proteins (e.g., amyloid-β, tau, alpha-synuclein) leads to neurodegeneration, synaptic dysfunction, and cognitive decline. | Impaired protein folding, decreased chaperone function (HSP70, HSP90), and defective autophagy. | Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, ALS. | [61] |
| Muscle cells | Decline in muscle function due to impaired protein turnover and aggregation of defective proteins such as in amyotrophic lateral sclerosis (ALS) and sarcopenia. | Decreased chaperone activity, proteasome dysfunction, and reduced autophagic activity. | Muscle atrophy, sarcopenia, ALS. | [63] |
| Heart | Proteostasis dysfunction impairs cardia c cells’ ability to remove misfolded proteins, leading to heart failure and cardiomyopathy. | Impaired protein quality control (UPS and autophagy), reduced mitochondrial function, and accumulation of protein aggregates. | Heart failure, cardiomyopathy, and arrhythmias. | [64] |
| Liver | Accumulation of misfolded proteins disrupts liver cell function, leading to liver diseases such as fatty liver and fibrosis. | Decline in autophagic processes, proteasomal activity, and overall protein degradation efficiency. | Non-alcoholic fatty liver disease (NAFLD), liver fibrosis. | [65] |
| Kidneys | Proteostasis decline in renal cells leads to the accumulation of toxic protein aggregates and kidney dysfunction. | Impaired autophagic clearance, decreased proteasomal function, and mitochondrial stress in renal cells. | Chronic kidney disease, renal fibrosis, and glomerulosclerosis. | [66] |
Proteostasis and age-related diseases: neurodegenerative diseases
Alzheimer’s disease (AD)
Proteostasis, the cellular process that ensures proper protein synthesis, folding, and degradation, is critical for maintaining cellular function (Figure 3). In the context of AD, disruptions in proteostasis contribute significantly to the progression of the disease. As AD is characterized by the accumulation of misfolded proteins, the breakdown of proteostasis exacerbates the buildup of these toxic aggregates, leading to neuronal dysfunction, synaptic loss, and cognitive decline. The primary misfolded proteins involved in AD are Aβ and tau, which, when not properly managed by proteostasis mechanisms, form damaging plaques and tangles in the brain [67, 68]. The process of protein folding in AD is disrupted, with amyloid-beta and tau failing to achieve proper conformations and instead aggregating into toxic oligomers, fibrils, and plaques. Amyloid-beta is generated from the amyloid precursor protein (APP) by the sequential cleavage of secretases, but in AD, this peptide accumulates due to inefficient clearance mechanisms. Similarly, tau, a protein involved in stabilizing microtubules, becomes hyperphosphorylated in AD, causing it to detach from microtubules and form neurofibrillary tangles inside neurons. These tangles disrupt essential neuronal functions such as intracellular transport and synaptic communication, contributing to cognitive impairment and neurodegeneration [69-71]. In animal models, impaired protein folding, aggregation, and degradation pathways have been shown to exacerbate the accumulation of toxic proteins such as amyloid-beta and tau, which are central to AD pathology. These animal studies have provided valuable insights into how proteostasis decline accelerates the development and progression of AD. Table 4 summarizes several animal studies that investigate the impact of proteostasis dysfunction on the progression of Alzheimer’s disease in aging.
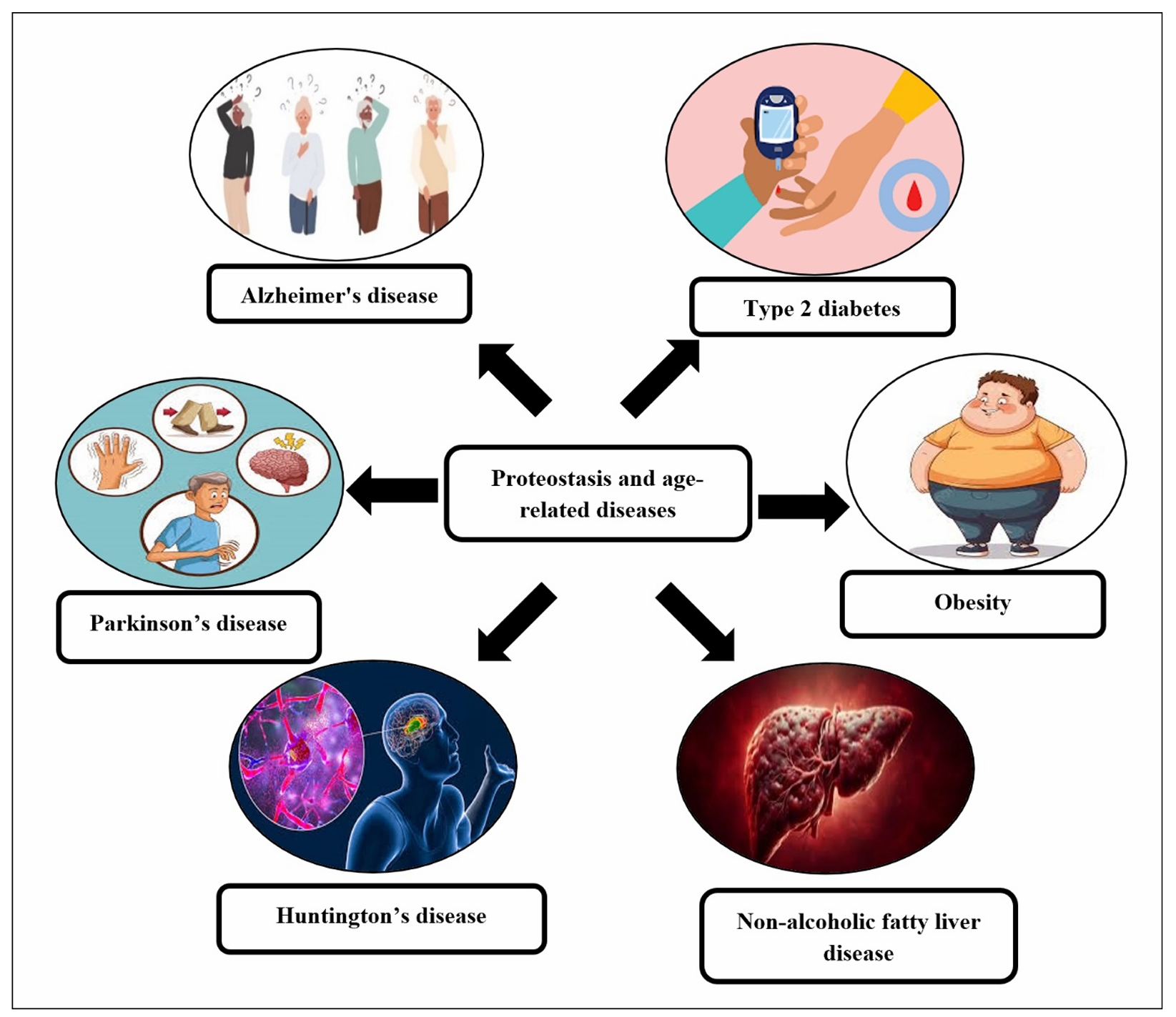
Figure 3. Proteostasis and age-related diseases.
Table 4.
Animal studies investigating the progression of Alzheimer’s disease (AD) due to proteostasis dysfunction in aging.
| Animal model | Proteostasis mechanism studied | Findings | References |
|---|---|---|---|
| Transgenic mice (3xTg-AD) | Ubiquitin-Proteasome System (UPS) | Aging worsens UPS dysfunction, increasing protein aggregation and neuroinflammation, leading to cognitive decline. | [81] |
| AβPP/PS1 transgenic mice | Autophagy | Impaired autophagy in aging promotes Aβ and tau accumulation, worsening memory. | [82] |
| AβPP/PS1 double transgenic mice | ER Stress (UPR) | Aging increases ER stress, leading to tau phosphorylation and neuronal damage. | [83] |
| Tau transgenic mice (rTg4510) | Chaperones (Hsp70, Hsp90) | Reduced chaperone levels in aging increase tau aggregation. | [84] |
| APP/PS1 transgenic mice | Mitochondrial Dysfunction | Mitochondrial dysfunction accelerates cognitive decline in aged mice. | [85] |
| Tg2576 mouse model | Autophagy and UPS | Impaired autophagy and UPS contribute to Aβ buildup and memory loss in aging. | [86] |
| 5xFAD transgenic mice | Proteasome and Autophagy Cross-talk | Failure of both UPS and autophagy accelerates Aβ accumulation and cognitive decline. | [87] |
| AppNL-F/NL-F mice | Chaperone-assisted protein degradation | Aging reduces chaperone activity, leading to increased Aβ aggregation. | [88] |
| 3xTg-AD mice | Proteasome Function | Aging impairs proteasomal degradation, leading to neurotoxic protein accumulation. | [89] |
| Tg2576 mice | ER Stress and UPR | Chronic ER stress in aging worsens Aβ pathology and cognitive decline. | [90] |
A critical aspect of proteostasis failure
in AD is the impairment of two major protein
degradation systems: the ubiquitin-proteasome
system (UPS) and autophagy. The UPS is responsible
for tagging misfolded proteins with ubiquitin and
directing them for degradation in the proteasome.
In AD, however, the efficiency of the UPS is reduced,
leading to the accumulation of damaged proteins like
amyloid-beta and tau. Similarly, autophagy, a process
that degrades damaged proteins and organelles in
lysosomes, is often impaired in AD, contributing to
the buildup of protein aggregates. The inability to
clear misfolded proteins accelerates neuronal damage,
making it a central feature in the progression of the
disease [72]. Molecular chaperones, such as Hsp70 and
Hsp90, are proteins that assist in the proper folding of
other proteins and prevent aggregation. However, in AD,
the function of these chaperones is compromised, which
exacerbates the accumulation of misfolded amyloid-beta and tau.
Chaperones play an essential role in maintaining proteostasis,
and when their activity is reduced, protein aggregation becomes
more likely, accelerating disease progression. The loss of
chaperone activity further compounds the problem, as it hampers
the cell’s ability to manage misfolded proteins, thus promoting
the accumulation of toxic aggregates [73-76].
In addition to these disruptions, endoplasmic reticulum (ER)
stress plays a significant role in AD. The ER is responsible
for protein folding and quality control, but when overwhelmed
by an excess of misfolded proteins like amyloid-beta, it activates
the unfolded protein response (UPR). The UPR aims to restore
proteostasis by halting protein synthesis and increasing protein
degradation. However, in AD, the UPR is often prolonged, leading
to neuronal dysfunction and death. Chronic ER stress can activate
apoptotic pathways, further exacerbating synaptic loss and
neurodegeneration. Proteostasis failure in AD also triggers an
inflammatory response in the brain. The accumulation of amyloid-beta
and tau aggregates activates glial cells, such as microglia and
astrocytes, which play a role in neuroinflammation. While this
response is intended to clear toxic proteins, chronic inflammation
worsens proteostasis dysfunction by inhibiting protein degradation
systems and promoting further protein aggregation. This inflammatory
feedback loop contributes to disease progression and worsens the overall
neuronal environment [77, 78]. Genetic and environmental factors also
influence proteostasis failure in AD. Certain mutations, such as those
in the APP gene or presenilins (PS1, PS2), lead to an overproduction of
amyloid-beta or impair its clearance, further disrupting proteostasis.
Environmental factors like oxidative stress, metabolic dysfunction,
and aging can also impair proteostasis mechanisms, making neurons
more vulnerable to the toxic effects of misfolded proteins.
Given the central role of proteostasis in AD, therapeutic
strategies targeting protein aggregation, folding, and
degradation pathways have emerged as potential treatments.
Enhancing the activity of the proteasome and autophagy systems
could help clear amyloid-beta and tau aggregates, potentially
slowing or halting disease progression. Additionally, immunotherapies
targeting amyloid-beta or tau have shown promise in reducing the
buildup of these toxic proteins. Another potential approach involves
boosting the activity of molecular chaperones to prevent the misfolding
and aggregation of proteins. Further research into restoring proteostasis
through these mechanisms offers hope for the development of disease-modifying
therapies for AD [79, 80].
Parkinson’s disease (PD)
In PD, proteostasis is compromised,
leading to the accumulation of misfolded proteins,
particularly α-synuclein, which aggregates into toxic
forms known as Lewy bodies. These aggregates disrupt
cellular functions, including synaptic activity,
mitochondrial function, and axonal transport,
contributing to the degeneration of dopaminergic
neurons in the substantia nigra, the brain region
most affected in PD. The failure of proteostasis
in PD is primarily driven by dysfunction in key
protein degradation pasthways, including the UPS
and autophagy. The UPS is responsible for tagging
misfolded or damaged proteins with ubiquitin,
marking them for degradation by the proteasome.
However, in PD, the efficiency of the UPS is often
impaired. This is particularly evident in cases where
mutations in the Parkin gene, which plays a key role
in protein degradation, prevent the efficient clearance
of damaged proteins. As a result, proteins like α-synuclein
accumulate in neurons, forming toxic aggregates that interfere
with normal cellular processes and contribute to neurodegeneration.
Similarly, the ALP, which is responsible for clearing larger
protein aggregates and damaged organelles, is also disrupted
in PD. Impairment of autophagy prevents the clearance of
α-synuclein and other misfolded proteins, accelerating the
buildup of toxic aggregates and promoting neuronal damage [91-95].
One of the most prominent features of PD pathology
is the aggregation of α-synuclein, a protein that under
normal conditions helps regulate neurotransmitter release
and synaptic function. However, in PD, α-synuclein becomes
misfolded and aggregates into insoluble fibrils, forming
Lewy bodies that are toxic to neurons. Normally, cellular
chaperones such as Hsp70 and Hsp90 help prevent α-synuclein
aggregation and facilitate its degradation. In PD, the
activity of these chaperones is often insufficient,
allowing α-synuclein to misfold and aggregate. These
aggregates not only disrupt cellular functions but also
spread from neuron to neuron, propagating the disease
throughout the brain in a characteristic manner [96, 97].
Mitochondrial dysfunction is another hallmark of PD, and
it is closely linked to proteostasis failure. Mitochondria
are responsible for generating energy and maintaining
cellular homeostasis, but they are also susceptible to
damage by misfolded proteins and oxidative stress.
In PD, defective mitophagy—an autophagic process that
specifically clears damaged mitochondria is a significant
contributor to the progression of the disease. Mutations
in genes like PINK1 and Parkin, which are involved in the
regulation of mitophagy, result in the accumulation of
dysfunctional mitochondria that increase oxidative stress
and exacerbate neurodegeneration [98].
The impairment of proteostasis also triggers
inflammation within the brain, further accelerating
PD progression. The accumulation of misfolded α-synuclein
aggregates activates microglia, the immune cells of the
brain, which release pro-inflammatory cytokines that
promote neuroinflammation. This inflammatory response
not only worsens neuronal damage but also impairs the
function of proteostasis systems, creating a vicious
cycle that accelerates the degeneration of dopaminergic
neurons. Chronic inflammation further exacerbates oxidative
stress and disrupts cellular proteostasis, making it more
difficult for neurons to clear misfolded proteins and damaged
organelles [99-102]. Genetic mutations contribute to proteostasis
failure in PD, particularly in familial forms of the disease.
Mutations in genes such as Parkin, PINK1, and DJ-1 impair the cellular
machinery responsible for protein degradation, making neurons more
susceptible to the accumulation of misfolded proteins like α-synuclein.
These mutations highlight the critical role of proteostasis in PD
and provide insights into the molecular mechanisms driving the disease.
Additionally, mutations in the α-synuclein gene itself can lead to an
increased propensity for misfolding and aggregation, further promoting
the accumulation of toxic proteins [103, 104]. Animal studies have
demonstrated that disruptions in protein quality control mechanisms,
such as impaired autophagy and proteasomal degradation, contribute to
the accumulation of misfolded proteins like alpha-synuclein, which is
a hallmark of PD pathology. These studies help elucidate how the decline
in proteostasis over time accelerates the onset and progression of Parkinson’s Disease (Table 5).
Table 5.
Animal studies investigating the progression of Parkinson’s disease (PD) due to proteostasis dysfunction in aging.
| Animal model | Proteostasis mechanism studied | Findings | Reference |
|---|---|---|---|
| α-synuclein transgenic mice | Ubiquitin-Proteasome System (UPS) | Aging leads to the accumulation of α-synuclein aggregates due to UPS dysfunction, contributing to neurodegeneration and motor deficits in PD. | [105] |
| MPTP-treated mice (Parkinson’s model) | Autophagy | Impaired autophagic degradation of misfolded proteins such as α-synuclein accelerates dopaminergic neurodegeneration in aging mice. | [106] |
| Park2 knockout mice (Parkinson’s model) | Parkin and Mitophagy | Aging exacerbates mitochondrial dysfunction and impairments in Parkin-mediated mitophagy, leading to dopaminergic degeneration and motor deficits. | [107] |
| α-synuclein transgenic mice | Chaperones (Hsp70, Hsp90) | Decreased levels of Hsp70 and Hsp90 with aging lead to the accumulation of α-synuclein aggregates, promoting neurodegeneration and motor impairments. | [108] |
| α-synuclein transgenic mice | Proteasome Function | Aging exacerbates proteasome dysfunction, leading to the accumulation of ubiquitinated α-synuclein and dopaminergic cell death. | [109] |
| α-synuclein transgenic mice | Autophagy and UPS | Both autophagy and UPS impairments in aging promote α-synuclein aggregation and neurodegeneration, contributing to PD pathogenesis. | [110] |
| MPTP-induced mice model | Mitochondrial Dysfunction | Aging increases mitochondrial dysfunction and decreases mitophagy, exacerbating dopaminergic cell death and motor deficits in PD. | [111] |
| Park2 mutant mice (Parkinson’s model) | Mitophagy | Defective mitophagy in aging leads to the accumulation of damaged mitochondria and dopaminergic neurodegeneration in PD models. | [112] |
| α-synuclein transgenic mice | Exosome-mediated protein degradation | Exosome secretion and the clearance of α-synuclein are impaired in aging, contributing to the accumulation of toxic aggregates and neurodegeneration. | [113] |
| α-synuclein transgenic mice | Chaperone-mediated autophagy (CMA) | CMA dysfunction in aging leads to α-synuclein aggregation and progressive dopaminergic neuronal death. | [114] |
Huntington’s disease (HD)
HD is a neurodegenerative disorder
caused by an expansion of the CAG
repeat in the HTT gene, which encodes
the protein huntingtin. In HD, the
polyglutamine (polyQ) tract within
huntingtin becomes abnormally long,
leading to the misfolding and aggregation
of huntingtin, which in turn disrupts
cellular proteostasis and contributes to
neuronal dysfunction and death. The
failure of proteostasis pathways,
including the UPS and autophagy, plays a
central role in HD progression by promoting
the accumulation of misfolded huntingtin and
other toxic proteins, thereby accelerating
neurodegeneration. One of the primary contributors
to proteostasis dysfunction in HD is the
accumulation of misfolded mutant huntingtin (mHTT),
which forms inclusions in neurons. These inclusions
are highly toxic and disrupt various cellular processes,
including vesicular trafficking, gene expression,
and mitochondrial function. Normally, the UPS is responsible
for tagging damaged or misfolded proteins with ubiquitin and
targeting them for degradation by the proteasome. However,
in HD, the UPS is overwhelmed by the accumulation of mHTT
aggregates, leading to impaired clearance of the toxic protein.
Studies have shown that the UPS is significantly impaired in HD,
as the presence of mHTT interferes with the function of the proteasome,
further exacerbating proteostasis failure and promoting the
accumulation of other misfolded proteins [115-117].
In addition to the UPS, autophagy, a critical
process that clears damaged proteins and organelles,
is also compromised in HD. The autophagy-lysosome
pathway (ALP), which is responsible for the degradation
of large protein aggregates and dysfunctional organelles,
is dysfunctional in HD. The presence of mHTT disrupts the
normal function of autophagy, preventing the efficient
clearance of aggregates. Autophagic impairment in HD
contributes to the buildup of toxic aggregates and
dysfunctional mitochondria, leading to increased
oxidative stress, cellular damage, and neuronal
death. Interestingly, enhancing autophagy has
been shown in some models of HD to improve the
clearance of mHTT aggregates and ameliorate
some aspects of the disease, suggesting that
restoring autophagic function may be a therapeutic
strategy. Moreover, the failure of molecular chaperones,
such as Hsp70 and Hsp90, which help in the proper folding
of proteins and prevent aggregation, also contributes to
proteostasis dysfunction in HD. In healthy cells, these
chaperones assist in refolding misfolded proteins or
targeting them for degradation. However, in HD, the
chaperone system is overwhelmed by the excess of misfolded mHTT,
reducing the ability of the cell to cope with protein misfolding.
This leads to an increased burden of protein aggregation, which
damages cellular components and accelerates the disease process.
Additionally, chaperones like Hsp70 can also help in clearing
mHTT aggregates via autophagy, making them crucial players in
maintaining proteostasis in HD [118-120].
The mitochondrial dysfunction observed in HD
is another key aspect of proteostasis failure.
Mitochondria are essential for energy production
and cellular homeostasis, and their function is
particularly important for neurons, which are
highly energy-demanding. mHTT aggregates impair
mitochondrial function by disrupting mitochondrial
dynamics, including fission, fusion, and transport,
and by increasing mitochondrial permeability,
leading to energy deficits and increased oxidative
stress. Both the UPS and autophagy are involved in
the clearance of damaged mitochondria, and their
dysfunction contributes to mitochondrial damage in HD,
exacerbating neurodegeneration. Impaired mitophagy,
the process by which damaged mitochondria are degraded,
has been observed in HD, further contributing to cellular
dysfunction and neuronal loss [121]. Chronic inflammation,
often observed in neurodegenerative diseases, also plays a
role in proteostasis dysfunction in HD. The accumulation of
mHTT and other misfolded proteins activates microglia, the
resident immune cells of the brain, leading to the release
of pro-inflammatory cytokines. This neuroinflammation can
further impair proteostasis by affecting protein degradation
pathways, such as the UPS and autophagy. Inflammatory
cytokines can also increase oxidative stress, which in turn
damages cellular components, including proteins, lipids, and
DNA, creating a vicious cycle of proteostasis failure and
neurodegeneration [122, 123]. Animal studies investigating HD
have shown that the accumulation of misfolded huntingtin protein,
a hallmark of the disease, is linked to impaired protein degradation
and aggregation pathways. These studies provide valuable insights
into how the decline of proteostasis contributes to the progression
of HD (Table 6).
Table 6.
Animal studies investigating the progression of Huntington’s disease (HD) due to proteostasis dysfunction in aging.
| Animal model | Proteostasis mechanism studied | Findings | Reference |
|---|---|---|---|
| Huntington’s disease knock-in mice (HDKI) | Ubiquitin-proteasome system (UPS) | Aging increases the accumulation of polyglutamine aggregates due to impaired UPS function, contributing to neuronal toxicity and motor deficits in HD. | [124] |
| R6/2 mice (HD model) | Autophagy | Autophagic dysfunction with age leads to the accumulation of Huntingtin aggregates, which contributes to neuronal degeneration and motor impairment in HD. | [125] |
| R6/2 transgenic mice | Proteasome function | Proteasome dysfunction in aging enhances the accumulation of Huntingtin aggregates, which leads to synaptic dysfunction and neuronal death. | [126] |
| R6/2 mice | Autophagy and the UPS | Defective autophagy and UPS function in aging contribute to the accumulation of toxic Huntingtin aggregates, leading to motor deficits and neurodegeneration. | [127] |
| ZQ175 mice (HD model) | Mitochondrial dysfunction | Aging leads to increased mitochondrial dysfunction, and impaired mitophagy, exacerbating the accumulation of Huntingtin aggregates and neuronal loss. | [128] |
| R6/2 transgenic mice | Protein aggregation | Aging accelerates Huntingtin protein aggregation, leading to neurodegeneration and motor dysfunction in HD models. | [129] |
| Q175 knock-in mice | Chaperone-mediated protein degradation | Age-related decline in Hsp70 function exacerbates Huntingtin inclusion formation, promoting progressive motor deficits and neurodegeneration. | [130] |
| HdhQ150 mice (HD model) | Autophagy and lysosomal pathways | Autophagy defects in aging lead to the accumulation of toxic Huntingtin aggregates, contributing to striatal neurodegeneration and movement disorders. | [131] |
Metabolic disorders
Disruption of proteostasis is implicated in the progression of age-related metabolic disorders, such as type 2 diabetes (T2D), obesity, and non-alcoholic fatty liver disease (NAFLD). These disorders are often characterized by the accumulation of misfolded proteins, mitochondrial dysfunction, and altered cellular signaling pathways, all of which are exacerbated by impaired proteostasis. In age-related metabolic diseases, proteostasis failure leads to dysfunction in key organs, including the liver, pancreas, and adipose tissue, which are critical for maintaining energy balance, glucose homeostasis, and lipid metabolism. Proteostasis dysfunction has been implicated in the progression of various metabolic disorders, including obesity, type 2 diabetes, insulin resistance, and fatty liver disease, particularly as aging compromises cellular quality control mechanisms. Animal studies have shown that the decline in protein folding, degradation, and recycling systems contributes to the development and exacerbation of these metabolic conditions. For example, impaired autophagy and proteasomal function in aging models lead to the accumulation of damaged proteins and organelles, disrupting metabolic homeostasis. Table 7 summarizes key animal studies that explore the relationship between proteostasis dysfunction and the progression of metabolic disorders in aging.
Table 7.
Animal studies investigating the progression of metabolic
disorders (such as obesity, type 2 diabetes, insulin
resistance, and fatty liver disease) due to proteostasis dysfunction in aging.
| Animal model | Proteostasis mechanism studied | Findings | Reference |
|---|---|---|---|
| C57BL/6J mice | Ubiquitin-proteasome system (UPS) | Aging impairs UPS function, leading to the accumulation of misfolded proteins, insulin resistance, and adiposity. | [136] |
| ApoE knockout mice | Autophagy | Age-related decline in autophagic activity exacerbates liver steatosis, insulin resistance, and obesity. | [137] |
| Db/db mice (type 2 diabetes model) | Endoplasmic reticulum stress (ER Stress) | Aging-induced ER stress leads to impaired insulin signaling, promoting type 2 diabetes and obesity. | [7] |
| C57BL/6 mice on high-fat diet | Mitochondrial dysfunction | Mitochondrial dysfunction during aging reduces energy expenditure and promotes insulin resistance and fatty liver. | [138] |
| Ob/Ob mice (obesity model) | Chaperones (Hsp70) | Decline in Hsp70 levels with aging leads to protein aggregation, impairing glucose metabolism and promoting obesity. | [139] |
| C57BL/6J mice | Proteostasis network (autophagy, UPS) | Impaired proteostasis network in aging leads to the accumulation of misfolded proteins in liver and adipose tissue, contributing to insulin resistance and fatty liver. | [140] |
| C57BL/6J mice (diet-induced obesity) | Autophagy and lipid metabolism | Aging-related autophagy defects contribute to lipid accumulation and insulin resistance, particularly in adipose tissue. | [141] |
| Zfp281 knockout mice | Chaperone-mediated protein degradation | Impaired chaperone-mediated protein degradation leads to glucose intolerance and fatty liver as mice age. | [142] |
| Sirt1 knockout mice | Proteostasis and mitochondrial quality control | Aging-related decline in Sirt1 reduces mitophagy, leading to insulin resistance and obesity in aging mice. | [143] |
| C57BL/6J mice (high-fat diet) | Lysosomal function and autophagy | Aging impairs lysosomal function and autophagic flux, contributing to obesity and insulin resistance in aging mice. | [144] |
Type 2 Diabetes (T2D): In T2D, proteostasis
failure is a key factor in the progression of the
disease. The pancreatic β-cells, which are responsible
for insulin secretion, are particularly vulnerable to
proteotoxic stress. The accumulation of misfolded or
aggregated proteins, particularly in the endoplasmic
reticulum (ER), disrupts normal cellular function and
induces ER stress. Under normal conditions, the ER is
responsible for protein folding, but under conditions
of metabolic stress (such as obesity and insulin
resistance), this process is overwhelmed. This leads
to the activation of the UPR, a cellular stress response
aimed at restoring proteostasis by enhancing protein
folding capacity and promoting degradation of misfolded
proteins. However, in the long term, persistent ER stress
can result in β-cell dysfunction and apoptosis,
contributing to impaired insulin secretion and the development of
insulin resistance. The UPS and autophagy also play significant
roles in maintaining proteostasis in T2D. In insulin-resistant
states, the accumulation of misfolded proteins and damaged
organelles (such as mitochondria) is observed, suggesting a
failure in both the UPS and autophagy. This accumulation of
damaged proteins disrupts insulin signaling and exacerbates
the systemic inflammation that contributes to insulin resistance.
Additionally, proteostasis failure in adipocytes (fat cells) can
affect adipose tissue function, leading to increased lipid accumulation
and the development of metabolic complications like dyslipidemia and
fatty liver [132, 133].
Obesity: In obesity, proteostasis failure plays a central
role in adipose tissue dysfunction, which contributes to the
progression of metabolic disorders. In the obese state, excessive
fat accumulation leads to an overload of proteins and lipids in adipocytes,
impairing normal protein folding and triggering ER stress. Chronic ER stress
in adipose tissue is linked to the development of insulin resistance and
inflammation, both of which contribute to the progression of obesity-related
metabolic disorders. The disruption of proteostasis in adipocytes also affects
adipokine production, including leptin and adiponectin, which are crucial for
regulating appetite, energy expenditure, and insulin sensitivity. Dysregulation
of these pathways leads to a vicious cycle of obesity and metabolic dysfunction.
Moreover, obesity-induced inflammation activates microglia and macrophages in adipose
tissue, further exacerbating proteostasis failure. These immune cells release pro-inflammatory
cytokines, which impair cellular functions in adipocytes and other tissues. Mitochondrial
dysfunction in adipocytes and other tissues, exacerbated by the accumulation of misfolded
proteins, also contributes to the progression of obesity and metabolic disease. Impaired
mitochondrial function leads to increased oxidative stress and inflammation, further
accelerating cellular damage and metabolic dysfunction [134, 135].
NAFLD: In NAFLD, proteostasis dysfunction contributes to the accumulation
of misfolded proteins in the liver, leading to hepatocyte stress and liver
damage. The liver plays a central role in regulating lipid metabolism,
detoxification, and protein synthesis. In NAFLD, excessive lipid accumulation,
particularly triglycerides, leads to cellular stress and the activation of the
UPR in the ER. The liver’s ability to handle this stress is compromised due to
impaired proteostasis, resulting in hepatocyte injury, inflammation, and fibrosis.
Chronic ER stress in liver cells is associated with insulin resistance, steatosis
(fatty liver), and steatohepatitis, the progression of which can eventually lead to
cirrhosis and liver failure. The UPS and autophagy are crucial for the clearance of
damaged proteins and organelles in hepatocytes. In NAFLD, these protein degradation
systems are often impaired, leading to the accumulation of damaged proteins, including
those involved in mitochondrial dysfunction. Mitochondria are essential for energy
production and maintaining metabolic homeostasis in the liver, and their dysfunction
is a hallmark of NAFLD. Impaired mitochondrial function further exacerbates oxidative
stress and the development of insulin resistance. Restoration of proteostasis in hepatocytes
through enhancing autophagy or the UPS could help prevent or slow the progression of NAFLD [135].
Therapeutic approaches in targeting proteostasis for healthy aging
Targeting chaperones and folding pathways
One promising therapeutic approach to maintain proteostasis in aging involves the development of chaperone-enhancing drugs. Molecular chaperones, like HSP70 and HSP90, play a crucial role in protein folding, stability, and preventing aggregation. In aging, these chaperones become less efficient, leading to the accumulation of misfolded proteins and cellular dysfunction. By enhancing the activity of these chaperones, it may be possible to improve protein quality control and slow down the progression of age-related diseases. Small molecules such as Geranylgeranyl acetone (GGA) have been shown to activate heat shock proteins and reduce neurodegenerative symptoms in animal models, particularly in Alzheimer’s and Parkinson’s diseases [145]. Moreover, the development of small molecule inhibitors that target the molecular chaperone Hsp90 has shown promise in preclinical trials for conditions like cancer and neurodegenerative diseases, suggesting that enhancing chaperone activity could help in ameliorating the proteostasis dysfunctions associated with aging [28]. However, more clinical trials and validation in human studies are needed to confirm their efficacy and safety for long-term use in aging populations [146].
Modulating proteolytic systems
Modulating the proteolytic systems, particularly the ubiquitin-proteasome system (UPS) and autophagy, is another approach to restore proteostasis. The UPS is responsible for degrading misfolded or damaged proteins, while autophagy helps in the removal of larger cellular debris, including damaged organelles. Both pathways deteriorate with age, contributing to the accumulation of toxic protein aggregates. Activating these pathways could clear accumulated misfolded proteins and prevent the cellular damage associated with aging and disease. Research has shown that enhancing UPS activity using small molecules like epoxomicin, a proteasome inhibitor, can improve protein degradation in aged cells, leading to enhanced protein homeostasis [147]. Similarly, boosting autophagy through compounds like rapamycin (which inhibits the mTOR pathway) has been linked to improved longevity and reduced age-related disease in model organisms [148]. These strategies, however, need to be carefully managed, as overactivation of proteolytic pathways could lead to cellular stress or undesirable effects. Clinical trials investigating autophagy enhancers and proteasome activators are underway as summarized in Table 8, but significant challenges remain in fine-tuning these pathways for therapeutic use [149].
Table 8.
Some ongoing clinical trials related to proteolysis in aging.
| Study name | Clinical trial ID | Intervention | Phase | Focus area | Status |
|---|---|---|---|---|---|
| Lomecel-B for aging frailty | NCT03735277 | Mesenchymal Stem Cells (MSCs) | Phase II | Aging frailty and sarcopenia | Active, recruiting |
| Senolytic therapy for aging | NCT03814783 | Dasatinib + Quercetin | Phase I/II | Cellular senescence and aging | Active, recruiting |
| Exosome therapy for aging frailty | NCT04162357 | Exosomes derived from MSCs | Phase I/II | Age-related frailty | Active, recruiting |
| Targeting senescence in cardiovascular aging | NCT04531443 | Navitoclax (Bcl-2 inhibitor) | Phase I | Cardiovascular aging and endothelial dysfunction | Recruiting |
| Proteostasis restoration in neurodegeneration | NCT04644313 | Proteostasis regulators (e.g., modafinil) | Phase I | Alzheimer’s and neurodegenerative diseases | Recruiting |
| Impact of autophagy enhancement on aging | NCT03063689 | Rapamycin | Phase II | Autophagy in aging-related conditions | Active, not recruiting |
Emerging therapeutics
Several promising proteostasis regulators
are currently in clinical trials, aiming
to slow aging and treat age-related diseases
by restoring proteostasis. One such approach
involves Nrf2 activators, which can regulate
the expression of genes involved in oxidative
stress response and protein degradation. Preclinical
studies have shown that activating Nrf2 can enhance
the cellular antioxidant capacity and improve proteostasis,
potentially delaying age-related neurodegeneration and
metabolic dysfunction [150]. Another class of proteostasis
regulators under investigation includes Hsp70 inducers like
BGP-15, which has been shown to promote protein refolding
and prevent aggregation in neurodegenerative diseases [151].
These compounds are undergoing early-phase clinical trials,
and while the results are promising, the long-term safety and
effectiveness of these therapies require further exploration.
A significant challenge in this area remains developing drugs
that can specifically target proteostasis pathways without
causing off-target effects, particularly when using systemic
activators that affect multiple tissues in the body.
Conclusions
This review underscores the pivotal role of
proteostasis in maintaining cellular function,
particularly in aging and neurodegenerative
diseases. Proteostasis, the balance of protein
synthesis, folding, and degradation, is maintained
by molecular chaperones like heat shock proteins
(HSPs), which ensure proper protein folding, prevent
aggregation, and promote the degradation of misfolded
proteins. Chaperones, including Hsp70, Hsp90, and small
heat shock proteins, are crucial for maintaining protein
stability, especially under stress conditions such as heat
or oxidative stress. These proteins also guide misfolded
proteins to degradation pathways, such as the ubiquitin-proteasome
system (UPS) and autophagy, to prevent the accumulation of toxic
aggregates. The UPS plays a central role in maintaining proteostasis
by selectively degrading damaged or unnecessary proteins through a
regulated process involving ubiquitination and proteasomal
degradation. Autophagy further supports proteostasis by
degrading damaged proteins and organelles through lysosomal
pathways. However, with aging, the efficiency of both the
UPS and autophagy declines, leading to an accumulation of
misfolded or aggregated proteins. These aggregates, which
are resistant to degradation, disrupt cellular function
and contribute to neurodegenerative diseases, including
Alzheimer’s, Parkinson’s, and Huntington’s diseases.
Proteostasis dysfunction is a
hallmark of aging, and as molecular
chaperones, proteolytic systems,
and stress response pathways become
less efficient over time, the cellular
machinery struggles to manage protein
quality control. This dysfunction
exacerbates protein misfolding and
aggregation, triggering cellular
stress responses like the UPR, which,
when chronically activated, leads to
cellular damage and dysfunction. The
accumulation of toxic protein aggregates
in neurons is particularly detrimental
due to their limited regenerative capacity,
contributing to the onset and progression
of neurodegenerative diseases.
Future research scope
A critical gap in current proteostasis
research lies in understanding how cellular
proteostasis mechanisms differ from those
at the organismal level, particularly in
the context of aging. While cellular proteostasis
focuses on the maintenance of protein homeostasis
within individual cells, the systemic effects of
aging introduce additional complexities, such as
inter-organ communication and the accumulation of
damaged proteins across tissues. Future research
should focus on elucidating how proteostasis networks
function at the organismal level, exploring how aging
in one tissue or organ may influence proteostasis in others.
This will provide a more comprehensive understanding
of systemic aging and help identify novel therapeutic
targets for age-related diseases like Alzheimer’s and Parkinson’s.
Integrating high-throughput proteomics
with emerging technologies such as single-cell
RNA sequencing and organ-on-a-chip models holds
significant potential for bridging these gaps.
High-throughput proteomics will enable the
identification of age-related changes in
protein dynamics, while single-cell RNA
sequencing will offer insights into cellular
heterogeneity and how different cell types
maintain proteostasis as they age. Moreover,
organ-on-a-chip technologies can simulate human
tissue responses to proteostasis imbalances in a
controlled environment, advancing the development
of more accurate models for human aging.
Personalized medicine approaches, incorporating
genetic, environmental, and proteomic data, will
also be essential in tailoring interventions to
maintain proteostasis at the individual level,
offering potential for more effective therapeutic
strategies for aging and age-related diseases.
Declarations
Author contributions
Gaurav N. Kasar: Conceptualization, Investigation, Writing original draft, Pooja B. Rasal: Conceptualization, Investigation, Writing original draft, Chandrashekhar D. Patil: Resources, Data curation, Visualization, Formal analysis, Sunil K. Mahajan: Resources, Data curation, Visualization, Formal analysis, Aman B. Upaganlawar: Resources, Data curation, Visualization, Formal analysis.
Conflicts of interest
The authors confirm that there are no known conflicts of interest.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Funding
None.
References
1. Klaips CL, Jayaraj GG, & Hartl FU. Pathways of cellular proteostasis in aging and disease. J Cell Biol, 2018, 217(1): 51-63. [Crossref]
2. Margulis B, Tsimokha A, Zubova S, & Guzhova I. Molecular chaperones and proteolytic machineries regulate protein homeostasis in aging. Cells, 2020, 9(5): 1308-1318. [Crossref]
3. Balchin D, Hayer-Hartl M, & Hartl FU. In vivo aspects of protein folding and quality control. Science, 2016, 353(6294): aac4354. [Crossref]
4. Hipp MS, Park SH, & Hartl FU. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol, 2014, 24(9): 506-514. [Crossref]
5. Jucker M, & Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature, 2013, 501(7465): 45-51. [Crossref]
6. Mbatha NA, Mushebenge AG-A, & Khathi A. Identification of putative causal relationships between blood-based biomarkers and prediabetes-induced senescence: a comprehensive review. Physiologia, 2024, 4(2): 149-181. [Crossref]
7. López-Otín C, Blasco MA, Partridge L, Serrano M, & Kroemer G. The hallmarks of aging. Cell, 2013, 153(6): 1194-1217. [Crossref]
8. Tang J-X, & Xiao F-H. Editorial: the regulation of proteostasis in aging. Frontiers in Cell and Developmental Biology, 2023, 11: 1221510. [Crossref]
9. Löw P. The role of ubiquitin-proteasome system in ageing. Gen Comp Endocrinol, 2011, 172(1): 39-43. [Crossref]
10. Hartl FU, & Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol, 2009, 16(6): 574-581. [Crossref]
11. Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol, 2013, 14(10): 630-642. [Crossref]
12. Papsdorf K, & Richter K. Protein folding, misfolding and quality control: the role of molecular chaperones. Essays Biochem, 2014, 56: 53-68. [Crossref]
13. Beard JR, Officer A, de Carvalho IA, Sadana R, Pot AM, Michel JP, et al. The world report on ageing and health: a policy framework for healthy ageing. Lancet, 2016, 387(10033): 2145-2154. [Crossref]
14. Hipp MS, Park SH, & Hartl FU. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol, 2014, 24(9): 506-514. [Crossref]
15. Broz P, & Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol, 2016, 16(7): 407-420. [Crossref]
16. Díaz-Villanueva JF, Díaz-Molina R, & García-González V. Protein folding and mechanisms of proteostasis. Int J Mol Sci , 2015, 16(8): 17193-17230. [Crossref]
17. Kampinga HH, & Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol, 2010, 11(8): 579-592. [Crossref]
18. Zhang WH, Koyuncu S, & Vilchez D. Insights into the links between proteostasis and aging from C. elegans. Front Aging, 2022, 3: 854157. [Crossref]
19. Wang X, Liu X., Song K., & Du L. An insight into the roles of ubiquitin-specific proteases in plants: development and growth, morphogenesis, and stress response. Front Plant Sci , 2024, 15: 1396634. [Crossref]
20. Komander D, & Rape M. The ubiquitin code. Annu Rev Biochem, 2012, 81: 203-229. [Crossref]
21. Zheng N, & Shabek N. Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem, 2017, 86: 129-157. [Crossref]
22. Heard DS, Tuttle CSL, Lautenschlager NT, & Maier AB. Repurposing proteostasis-modifying drugs to prevent or treat age-related dementia: a systematic review. Front Physiol, 2018, 9: 1520-1530. [Crossref]
23. Vilchez D, Saez I, & Dillin A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat Commun, 2014, 5: 5659-5661. [Crossref]
24. Glickman MH, & Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev, 2002, 82(2): 373-428. [Crossref]
25. Berner N, Reutter KR, & Wolf DH. Protein quality control of the endoplasmic reticulum and ubiquitin-proteasome-triggered degradation of aberrant proteins: yeast pioneers the Path. Annu Rev Biochem, 2018, 87: 751-782. [Crossref]
26. Chen D, & Dou QP. The ubiquitin-proteasome system as a prospective molecular target for cancer treatment and prevention. Curr Protein Pept Sci, 2010, 11(6): 459-470. [Crossref]
27. Saez I, & Vilchez D. The mechanistic links between proteasome activity, aging and age-related diseases. Curr Genomics, 2014, 15(1): 38-51. [Crossref]
28. Kaushik S, & Cuervo AM. Proteostasis and aging. Nat Med, 2015, 21(12): 1406-1415. [Crossref]
29. Rubinsztein DC, Mariño G, & Kroemer G. Autophagy and aging. Cell, 2011, 146(5): 682-695. [Crossref]
30. López-Otín C, Galluzzi L, Freije JMP, Madeo F, & Kroemer G. Metabolic control of longevity. Cell, 2016, 166(4): 802-821. [Crossref]
31. Labbadia J, & Morimoto RI. Repression of the heat shock response is a programmed event at the onset of reproduction. Mol Cell, 2015, 59(4): 639-650. [Crossref]
32. Cuervo AM, & Dice JF. When lysosomes get old. Exp Gerontol, 2000, 35(2): 119-131. [Crossref]
33. Sala AJ, & Morimoto RI. Protecting the future: balancing proteostasis for reproduction. Trends Cell Biol, 2022, 32(3): 202-215. [Crossref]
34. Parzych KR, & Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal, 2014, 20(3): 460-473. [Crossref]
35. Yang Z, & Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol, 2010, 12(9): 814-822. [Crossref]
36. Quirós PM, Langer T, & López-Otín C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol, 2015, 16(6): 345-359. [Crossref]
37. Vilchez D, Simic MS, & Dillin A. Proteostasis and aging of stem cells. Trends Cell Biol, 2014, 24(3): 161-170. [Crossref]
38. Stoka V, Turk V, & Turk B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res Rev, 2016, 32: 22-37. [Crossref]
39. Freitas-Rodríguez S, Folgueras AR, & López-Otín C. The role of matrix metalloproteinases in aging: Tissue remodeling and beyond. Biochim Biophys Acta Mol Cell Res, 2017, 1864(11 Pt A): 2015-2025. [Crossref]
40. Bello AI, Goswami R, Brown SL, Costanzo K, Shores T, Allan S, et al. Deubiquitinases in Neurodegeneration. Cells, 2022, 11(3): 556-566. [Crossref]
41. Taylor J, & Ikeda M. Role of caspases in neuronal damage. Drug News Perspect, 2000, 13(1): 5-11.
42. Feng Y, Nouri K, & Schimmer AD. Mitochondrial ATP-dependent proteases-biological function and potential anti-cancer targets. Cancers, 2021, 13(9): 2020-2031. [Crossref]
43. Brancolini C, & Iuliano L. Proteotoxic stress and cell death in cancer cells. Cancer, 2020, 12(9): 2385-2395. [Crossref]
44. Boas SM, Joyce KL, & Cowell RM. The NRF2-dependent transcriptional regulation of antioxidant defense pathways: relevance for cell type-specific vulnerability to neurodegeneration and therapeutic intervention. Antioxidants, 2021, 11(1): 8-18. [Crossref]
45. Lee JM, & Johnson JA. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J Biochem Mol Biol, 2004, 37(2): 139-143. [Crossref]
46. Scheper W, & Hoozemans JJ. The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol, 2015, 130(3): 315-331. [Crossref]
47. Webb AE, & Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci, 2014, 39(4): 159-169. [Crossref]
48. Morimoto RI. Cell-nonautonomous regulation of proteostasis in aging and disease. Cold Spring Harb Perspect Biol, 2020, 12(4): a034074. [Crossref]
49. Labbadia J, & Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem, 2015, 84: 435-464. [Crossref]
50. Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol, 2013, 53: 401-426. [Crossref]
51. Lu MC, Ji JA, Jiang ZY, & You QD. The Keap1-Nrf2-ARE pathway as a potential preventive and therapeutic target: an update. Med Res Rev, 2016, 36(5): 924-963. [Crossref]
52. Ghemrawi R, & Khair M. Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseases. Int J Mol Sci, 2020, 21(17): 6127-6137. [Crossref]
53. Calderwood SK, Murshid A, & Prince T. The shock of aging: molecular chaperones and the heat shock response in longevity and aging--a mini-review. Gerontology, 2009, 55(5): 550-558. [Crossref]
54. Du S, & Zheng H. Role of FoxO transcription factors in aging and age-related metabolic and neurodegenerative diseases. Cell Biosci, 2021, 11(1): 188-198. [Crossref]
55. Francisco S, Martinho V, Ferreira M, Reis A, Moura G, Soares AR, et al. The role of microRNAs in proteostasis decline and protein aggregation during brain and skeletal muscle aging. International Journal of Molecular Sciences, 2022, 23(6): 3232-3243.
56. Soti C, & Csermely P. Aging and molecular chaperones. Exp Gerontol, 2003, 38(10): 1037-1040. [Crossref]
57. van Ziel AM, & Scheper W. The UPR in neurodegenerative disease: not just an inside job. Biomolecules, 2020, 10(8): 1090-1100. [Crossref]
58. Sonninen TM, Goldsteins G, Laham-Karam N, Koistinaho J, & Lehtonen Š. Proteostasis disturbances and inflammation in neurodegenerative diseases. Cells, 2020, 9(10): 2183-2193. [Crossref]
59. Watanabe Y, Taguchi K, & Tanaka M. Ubiquitin, autophagy and neurodegenerative diseases. Cells, 2020, 9(9): 2022-2032. [Crossref]
60. Momtaz S, Memariani Z, El-Senduny FF, Sanadgol N, Golab F, Katebi M, et al. Targeting ubiquitin-proteasome pathway by natural products: novel therapeutic strategy for treatment of neurodegenerative diseases. Front Physiol, 2020, 11: 361-371. [Crossref]
61. Pagan LU, Gomes MJ, Gatto M, Mota GAF, Okoshi K, & Okoshi MP. The role of oxidative stress in the aging heart. Antioxidants, 2022, 11(2): 336-346. [Crossref]
62. Guo J, Huang X, Dou L, Yan M, Shen T, Tang W, et al. Aging and aging-related diseases: from molecular mechanisms to interventions and treatments. Signal Transduct Target Ther, 2022, 7(1): 391-405. [Crossref]
63. Inagi R, Ishimoto Y, & Nangaku M. Proteostasis in endoplasmic reticulum--new mechanisms in kidney disease. Nat Rev Nephrol, 2014, 10(7): 369-378. [Crossref]
64. Hardy J, & Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science, 2002, 297(5580): 353-356. [Crossref]
65. Marino Gammazza A, Bavisotto CC, Barone R, de Macario EC, & Macario AJ. Alzheimer’s disease and molecular chaperones: current knowledge and the future of chaperonotherapy. Curr Pharm Des, 2016, 22(26): 4040-4049. [Crossref]
66. Dantuma NP, & Bott LC. The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front Mol Neurosci, 2014, 7: 70-81. [Crossref]
67. Almeida CG, Takahashi RH, & Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci, 2006, 26(16): 4277-4288. [Crossref]
68. Bourdenx M, Gavathiotis E, & Cuervo AM. Chaperone-mediated autophagy: a gatekeeper of neuronal proteostasis. Autophagy, 2021, 17(8): 2040-2042. [Crossref]
69. Moreira PI, Zhu X, Wang X, Lee HG, Nunomura A, Petersen RB, et al. Mitochondria: a therapeutic target in neurodegeneration. Biochim Biophys Acta, 2010, 1802(1): 212-220. [Crossref]
70. Rawat P, Sehar U, Bisht J, Selman A, Culberson J, & Reddy PH. Phosphorylated tau in Alzheimer’s disease and other tauopathies. Int J Mol Sci, 2022, 23(21): 12841. [Crossref]
71. Yerbury JJ, Ooi L, Dillin A, Saunders DN, Hatters DM, Beart PM, et al. Walking the tightrope: proteostasis and neurodegenerative disease. J Neurochem, 2016, 137(4): 489-505. [Crossref]
72. Ciechanover A, & Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med, 2015, 47(3): e147. [Crossref]
73. Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep, 2014, 9(3): 1135-1150. [Crossref]
74. Götz J, & Ittner LM. Animal models of Alzheimer’s disease and frontotemporal dementia. Nat Rev Neurosci, 2008, 9(7): 532-544. [Crossref]
75. Zhang Z, Yang X, Song YQ, & Tu J. Autophagy in Alzheimer’s disease pathogenesis: Therapeutic potential and future perspectives. Ageing Res Rev, 2021, 72: 101464. [Crossref]
76. El Ayadi A, Stieren ES, Barral JM, & Boehning D. Ubiquilin-1 regulates amyloid precursor protein maturation and degradation by stimulating K63-linked polyubiquitination of lysine 688. Proc Natl Acad Sci USA, 2012, 109(33): 13416-13421. [Crossref]
77. Penke B, Szűcs M, & Bogár F. New pathways identify novel drug targets for the prevention and treatment of Alzheimer’s disease. Int J Mol Sci, 2023, 24(6): 5383-5393. [Crossref]
78. Li YY, Qin ZH, & Sheng R. The multiple roles of autophagy in neural function and diseases. Neurosci Bull, 2024, 40(3): 363-382. [Crossref]
79. Martinez-Lopez N, Athonvarangkul D, & Singh R. Autophagy and aging. Adv Exp Med Biol, 2015, 847: 73-87. [Crossref]
80. Nikoletopoulou V, Papandreou ME, & Tavernarakis N. Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ, 2015, 22(3): 398-407. [Crossref]
81. Deng Z, Dong Y, Zhou X, Lu JH, & Yue Z. Pharmacological modulation of autophagy for Alzheimer’s disease therapy: opportunities and obstacles. Acta Pharm Sin B, 2022, 12(4): 1688-1706. [Crossref]
82. Ondaro J, Hernandez-Eguiazu H, Garciandia-Arcelus M, Loera-Valencia R, Rodriguez-Gómez L, Jiménez-Zúñiga A, et al. Defects of nutrient signaling and autophagy in neurodegeneration. Front Cell Dev Biol, 2022, 10: 836196. [Crossref]
83. Zhang K, Zhu S, Li J, Jiang T, Feng L, Pei J, et al. Targeting autophagy using small-molecule compounds to improve potential therapy of Parkinson’s disease. Acta Pharm Sin B, 2021, 11(10): 3015-3034. [Crossref]
84. Di Meco A, Curtis ME, Lauretti E, & Praticò D. Autophagy dysfunction in Alzheimer’s disease: mechanistic insights and new therapeutic opportunities. Biol Psychiatry, 2020, 87(9): 797-807. [Crossref]
85. Ratan Y, Rajput A, Maleysm S, Pareek A, Jain V, Pareek A, et al. An insight into cellular and molecular mechanisms underlying the pathogenesis of neurodegeneration in Alzheimer’s disease. Biomedicines, 2023, 11(5): 1398-1401. [Crossref]
86. Sin O, & Nollen EA. Regulation of protein homeostasis in neurodegenerative diseases: the role of coding and non-coding genes. Cell Mol Life Sci, 2015, 72(21): 4027-4047. [Crossref]
87. Kwon S, Iba M, Kim C, & Masliah E. Immunotherapies for aging-related neurodegenerative diseases-emerging perspectives and new targets. Neurotherapeutics, 2020, 17(3): 935-954. [Crossref]
88. Moya-Alvarado G, Gershoni-Emek N, Perlson E, & Bronfman FC. Neurodegeneration and Alzheimer’s disease (AD). What can proteomics tell us about the Alzheimer’s brain? Mol Cell Proteomics, 2016, 15(2): 409-425. [Crossref]
89. Nedelsky NB, Todd PK, & Taylor JP. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim Biophys Acta, 2008, 1782(12): 691-699. [Crossref]
90. Eshraghi M, Adlimoghaddam A, Mahmoodzadeh A, Sharifzad F, Yasavoli-Sharahi H, Lorzadeh S, et al. Alzheimer’s disease pathogenesis: role of autophagy and mitophagy focusing in microglia. Int J Mol Sci, 2021, 22(7): 3330-3340. [Crossref]
91. Andronie-Cioara FL, Ardelean AI, Nistor-Cseppento CD, Jurcau A, Jurcau MC, Pascalau N, et al. Molecular mechanisms of neuroinflammation in aging and Alzheimer’s disease progression. Int J Mol Sci, 2023, 24(3): 1869-1879. [Crossref]
92. Zhao Y, Lin M, Zhai F, Chen J, & Jin X. Exploring the role of ubiquitin-proteasome system in the pathogenesis of Parkinson’s disease. Pharmaceuticals, 2024, 17(6): 782-792. [Crossref]
93. Radanović T, & Ernst R. The unfolded protein response as a guardian of the secretory pathway. Cells, 2021, 10(11): 2965-2975. [Crossref]
94. Kurtishi A, Rosen B, Patil KS, Alves GW, & Møller SG. Cellular proteostasis in neurodegeneration. Mol Neurobiol, 2019, 56(5): 3676-3689. [Crossref]
95. Lane CA, Hardy J, & Schott JM. Alzheimer’s disease. Eur J Neurol, 2018, 25(1): 59-70. [Crossref]
96. Bobori C, Theocharopoulou G, & Vlamos P. Molecular chaperones in neurodegenerative diseases: a short review. Adv Exp Med Biol, 2017, 987: 219-231. [Crossref]
97. Ashraf GM, Greig NH, Khan TA, Hassan I, Tabrez S, Shakil S, et al. Protein misfolding and aggregation in Alzheimer’s disease and type 2 diabetes mellitus. CNS Neurol Disord Drug Targets, 2014, 13(7): 1280-1293. [Crossref]
98. Bonam SR, Tranchant C, & Muller S. Autophagy-lysosomal pathway as potential therapeutic target in Parkinson’s disease. Cells, 2021, 10(12): 3547-3557. [Crossref]
99. Day JO, & Mullin S. The genetics of Parkinson’s disease and implications for clinical practice. Genes, 2021, 12(7): 1006-1016. [Crossref]
100. Elsasser S, & Finley D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol, 2005, 7(8): 742-749. [Crossref]
101. Lotharius J, & Brundin P. Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci, 2002, 3(12): 932-942. [Crossref]
102. Dehay B, Bourdenx M, Gorry P, Przedborski S, Vila M, Hunot S, et al. Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol, 2015, 14(8): 855-866. [Crossref]
103. Lim KL. Ubiquitin-proteasome system dysfunction in Parkinson’s disease: current evidence and controversies. Expert Rev Proteomics, 2007, 4(6): 769-781. [Crossref]
104. Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, et al. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol, 2010, 67(12): 1464-1472. [Crossref]
105. Berthet A, Bezard E, Porras G, Fasano S, Barroso-Chinea P, Dehay B, et al. L-DOPA impairs proteasome activity in parkinsonism through D1 dopamine receptor. J Neurosci, 2012, 32(2): 681-691. [Crossref]
106. Blandini F, Sinforiani E, Pacchetti C, Samuele A, Bazzini E, Zangaglia R, et al. Peripheral proteasome and caspase activity in Parkinson disease and Alzheimer disease. Neurology, 2006, 66(4): 529-534. [Crossref]
107. Banerjee R, Beal MF, & Thomas B. Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci, 2010, 33(12): 541-549. [Crossref]
108. Malpartida AB, Williamson M, Narendra DP, Wade-Martins R, & Ryan BJ. Mitochondrial dysfunction and mitophagy in Parkinson’s disease: from mechanism to therapy. Trends Biochem Sci, 2021, 46(4): 329-343. [Crossref]
109. Jin SM, & Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci, 2012, 125(Pt 4): 795-799. [Crossref]
110. Stefanis L. α-Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med, 2012, 2(2): a009399. [Crossref]
111. Lee J, Sung KW, Bae EJ, Yoon D, Kim D, Lee JS, et al. 111 Targeted degradation of ⍺-synuclein aggregates in Parkinson’s disease using the AUTOTAC technology. Mol Neurodegener, 2023, 18(1): 41-51. [Crossref]
112. Tong H, Yang T, Xu S, Li X, Liu L, Zhou G, et al. Huntington’s disease: complex pathogenesis and therapeutic strategies. Int J Mol Sci, 2024, 25(7): 3845-3855. [Crossref]
113. Estevez-Fraga C, Flower MD, & Tabrizi SJ. Therapeutic strategies for Huntington’s disease. Curr Opin Neurol, 2020, 33(4): 508-518. [Crossref]
114. Ortega Z, & Lucas JJ. Ubiquitin-proteasome system involvement in Huntington’s disease. Front Mol Neurosci, 2014, 7: 77-87. [Crossref]
115. Ciechanover A, & Kwon YT. Protein quality control by molecular chaperones in neurodegeneration. Front Neurosci, 2017, 11: 185-195. [Crossref]
116. Sarkar S, & Rubinsztein DC. Huntington’s disease: degradation of mutant huntingtin by autophagy. Febs J, 2008, 275(17): 4263-4270. [Crossref]
117. Qi L, Zhang XD, Wu JC, Lin F, Wang J, DiFiglia M, et al. The role of chaperone-mediated autophagy in huntingtin degradation. PLoS One, 2012, 7(10): e46834. [Crossref]
118. Li XJ, & Li S. Proteasomal dysfunction in aging and Huntington disease. Neurobiol Dis, 2011, 43(1): 4-8. [Crossref]
119. Sweeney P, Park H, Baumann M, Dunlop J, Frydman J, Kopito R, et al. Protein misfolding in neurodegenerative diseases: implications and strategies. Transl Neurodegener, 2017, 6: 6-16. [Crossref]
120. Ross CA, & Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med, 2004, 10 Suppl: S10-17. [Crossref]
121. Bence NF, Sampat RM, & Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science, 2001, 292(5521): 1552-1555. [Crossref]
122. Ciechanover A, & Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron, 2003, 40(2): 427-446. [Crossref]
123. Bett JS, Goellner GM, Woodman B, Pratt G, Rechsteiner M, & Bates GP. Proteasome impairment does not contribute to pathogenesis in R6/2 Huntington’s disease mice: exclusion of proteasome activator REGgamma as a therapeutic target. Hum Mol Genet, 2006, 15(1): 33-44. [Crossref]
124. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet, 2004, 36(6): 585-595. [Crossref]
125. Cen X, Zhang M, Zhou M, Ye L, & Xia H. Mitophagy regulates neurodegenerative diseases. Cells, 2021, 10(8): 1876-1886. [Crossref]
126. Warrick JM, Chan HYE, Gray-Board GL, Chai Y, Paulson HL, & Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nature Genetics, 1999, 23(4): 425-428. [Crossref]
127. Gusella JF, MacDonald ME, Ambrose CM, & Duyao MP. Molecular genetics of Huntington’s disease. Arch Neurol, 1993, 50(11): 1157-1163. [Crossref]
128. Guo F, Liu X, Cai H, & Le W. Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol, 2018, 28(1): 3-13. [Crossref]
129. Hetz C, & Papa FR. The unfolded protein response and cell fate control. Mol Cell, 2018, 69(2): 169-181. [Crossref]
130. Kim YC, & Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest, 2015, 125(1): 25-32. [Crossref]
131. James HA, O’Neill BT, & Nair KS. Insulin regulation of proteostasis and clinical implications. Cell Metab, 2017, 26(2): 310-323. [Crossref]
132. Rocchi A, & He C. Emerging roles of autophagy in metabolism and metabolic disorders. Front Biol, 2015, 10(2): 154-164. [Crossref]
133. Sun-Wang JL, Yarritu-Gallego A, Ivanova S, & Zorzano A. The ubiquitin-proteasome system and autophagy: self-digestion for metabolic health. Trends Endocrinol Metab, 2021, 32(8): 594-608. [Crossref]
134. Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov, 2014, 4(8): 914-927. [Crossref]
135. Ghemrawi R, Battaglia-Hsu SF, & Arnold C. Endoplasmic reticulum stress in metabolic disorders. Cells, 2018, 7(6): 63-73. [Crossref]
136. Prasun P. Mitochondrial dysfunction in metabolic syndrome. Biochim Biophys Acta Mol Basis Dis, 2020, 1866(10): 165838. [Crossref]
137. Cuervo AM, & Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem, 2000, 275(40): 31505-31513. [Crossref]
138. Clemente-Postigo M, Tinahones A, El Bekay R, Malagón MM, & Tinahones FJ. The role of autophagy in white adipose tissue function: implications for metabolic health. Metabolites, 2020, 10(5): 179-189. [Crossref]
139. Massey AC, Zhang C, & Cuervo AM. Chaperone-mediated autophagy in aging and disease. Curr Top Dev Biol, 2006, 73: 205-235. [Crossref]
140. You Y, & Liang W. SIRT1 and SIRT6: The role in aging-related diseases. Biochim Biophys Acta Mol Basis Dis, 2023, 1869(7): 166815. [Crossref]
141. Guerrero-Navarro L, Jansen-Dürr P, & Cavinato M. Age-related lysosomal dysfunctions. Cells, 2022, 11(12): 1977-1987. [Crossref]
142. Uchida S, Fujiki M, Nagai Y, Abe T, & Kobayashi H. Geranylgeranylacetone, a noninvasive heat shock protein inducer, induces protein kinase C and leads to neuroprotection against cerebral infarction in rats. Neuroscience Letters, 2006, 396(3): 220-224. [Crossref]
143. Li ZN, & Luo Y. HSP90 inhibitors and cancer: prospects for use in targeted therapies (review). Oncol Rep, 2023, 49(1): 8443. [Crossref]
144. Beretta G, & Shala AL. Impact of heat shock proteins in neurodegeneration: possible therapeutical targets. Ann Neurosci, 2022, 29(1): 71-82. [Crossref]
145. Hakim V, Cohen LD, Zuchman R, Ziv T, & Ziv NE. The effects of proteasomal inhibition on synaptic proteostasis. Embo J, 2016, 35(20): 2238-2262. [Crossref]
146. Federico M, De la Fuente S, Palomeque J, & Sheu SS. The role of mitochondria in metabolic disease: a special emphasis on heart dysfunction. J Physiol, 2021, 599(14): 3477-3493. [Crossref]
147. Wang CH, & Wei YH. Roles of mitochondrial sirtuins in mitochondrial function, redox homeostasis, insulin resistance and type 2 diabetes. Int J Mol Sci, 2020, 21(15): 5266-5276. [Crossref]
148. Theurey P, & Pizzo P. The aging mitochondria. Genes, 2018, 9(1): 22-32. [Crossref]
149. Loeffler DA. Influence of normal aging on brain autophagy: a complex scenario. Frontiers in Aging Neuroscience, 2019, 11: 49-59. [Crossref]
150. Bustamante-Barrientos FA, Luque-Campos N, Araya MJ, Lara-Barba E, de Solminihac J, Pradenas C, et al. Mitochondrial dysfunction in neurodegenerative disorders: Potential therapeutic application of mitochondrial transfer to central nervous system-residing cells. J Transl Med, 2023, 21(1): 613-623. [Crossref]
151. Levine B, & Kroemer G. Autophagy in the pathogenesis of disease. Cell, 2008, 132(1): 27-42. [Crossref]