Open Access | Research
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
From plants to potential therapeutics: exploring neuroprotective properties against Alzheimer’s disease through molecular docking and MD simulations
* Corresponding author: Vikas Kumar
Mailing address: University Institute of Biotechnology, Chandigarh University, Gharuan, Mohali, Punjab, 140413, India.
Email: vikaskmr59@gmail.com
* Corresponding author: Ashwani Kumar
Mailing address: University Institute of Biotechnology, Chandigarh University, Gharuan, Mohali, Punjab, 140413, India.
Email: ashwanibitian@gmail.com
DOI: 10.31491/APT.2025.06.173
Abstract
Background: Alzheimer’s disease (AD) is a major global health
concern, characterized by the accumulation of abnormal protein aggregates that
cause cognitive decline. This study explores bioactive compounds from traditional
medicinal plants as potential therapeutic candidates for AD treatment.
Methods: A total of 39 phytochemicals from Withania somnifera,
Bacopa monnieri, Centella asiatica, and Crocus sativus were investigated for their
binding potential with AD-related enzymes, Acetylcholinesterase (AChE, PDB ID: 1B41)
and β-Secretase (BACE-1, PDB ID: 1TQF), using molecular docking and molecular dynamics
simulations. These compounds were further evaluated for drug-likeness and toxicity
prediction.
Results: Retinoic acid (-9.2 kcal/mol) and Somniferine (-8.8 kcal/mol)
demonstrated strong binding affinities with the target enzymes, as confirmed by
molecular docking. Molecular dynamics simulations further validated the stability
of these interactions. Additionally, drug-likeness and toxicity assessments
highlighted the therapeutic potential of these compounds.
Conclusion: This study identifies Retinoic acid as a promising
inhibitor of AChE and Somniferine as a novel inhibitor of BACE-1, suggesting
their potential for treating Alzheimer’s disease. Further in-vivo studies and
clinical trials are recommended to confirm their efficacy and therapeutic application.
Keywords
Alzheimer’s disease, bioactive compounds, acetylcholinesterase, beta-secretase, molecular docking, molecular dynamics; toxicity
Introduction
Alzheimer’s disease, a devastating neurodegenerative disorder, has emerged as a global
healthcare challenge. At present, dementia affects more than 55 million people globally,
with over 60% residing in low- and middle-income nations. Each year, nearly 10 million
new cases emerge. Alzheimer’s disease constitutes the most prevalent form of dementia,
accounting for 60–70% of diagnoses [1]. The complex pathophysiology of Alzheimer’s
disease is marked by the accumulation of abnormal protein aggregates in the brain,
including beta-amyloid plaques [2] and tau fibrillary tangles [3, 4] which ultimately
results in cognitive decline and memory impairment [5]. Alzheimer’s disease exhibits
two neuropathies: Positive Lesions and Negative Lesions [6]. Positive Lesions, caused
due to aggregation, include pathological changes like the formation of amyloid plaques
and neurofibril tangles. Negative Lesions, caused by deterioration, are characterized
by large atrophy (shrinking of the brain) due to neural and synaptic losses [7, 8].
Plaques are the accumulation of beta-amyloid (Aβ) proteins [9] around the neurons.
These Aβ deposits are formed by the catalytic action of β-secretase (BACE-1) [10]
and γ-secretase enzymes [11], causing the cleaving of transmembrane amyloid precursor
proteins (APP) [12]. APPs on cleavage form multiple insoluble Aβ monomers or peptides,
which are sticky and adhere to form plaques between the neurons [13]. This leads to an
interruption of neuron-to-neuron signaling, amyloid angiopathy [14] and inflammation
in the brain producing neurotoxic [15] effects and hence, cognitive impairments [16].
Neuro-fibrillary tangles (NFTs) are the intracellular aggregation of abnormal,
hyper-phosphorylated tau proteins [17]. Phosphate kinase gets activated due to a cascade
of reactions initiated by beta-amyloid plaques and transfers the phosphate group to
tau proteins causing their misfolding. Structurally altered tau proteins can no longer
support microtubules and start forming tangles in the neuro-cytoplasm, axons and dendrites.
This alteration in neurons sends the signal for programmed cell death and nerve cells
start undergoing apoptosis leading to the shrinkage of the brain [18]. The atrophy of
brain cells causes memory loss, language impairment, loss of motor skills, and
disorientation and ultimately makes the person bed-ridden leading to death [16, 19].
Despite significant research endeavors, the quest for effective therapies for Alzheimer’s
disease continues to be challenging.
Inspiration from nature has prompted exploration into potential therapies for Alzheimer’s
disease [20], leading to a focus on the neuroprotective medicinal plants Withania somnifera,
Bacopa monnieri, Centella asiatica, and Crocus sativus [21]. These plants have a rich
history of traditional use, with various parts of the plants believed to possess medicinal
properties. Withania somnifera (also known as Ashwagandha), a member of Solanaceae family
is known to possess neuroprotective [22, 23], hepatoprotective [24], anti-inflammatory,
antioxidant [25] and anti-depressant [26] properties. Bacopa monnieri (family: Scrophulariaceae)
also called Brahmi, has been used for years as a brain-tonic in Ayurveda [27], known for treating
various neurological disorders such as improving memory, thinking skills, insomnia, seizures,
and anxiety [28]. It has also been studied for its therapeutic potential to treat or prevent
neurodegenerative diseases like Alzheimer’s [29] and Parkinson’s disease [30]. Centella asiatica
from Apiaceae family, is another medicinally important [31] plant with antioxidant [32],
antimicrobial [33], anticancer [34], neuroceutical and cogniceutical properties [35, 36].
It is known to reduce oxidative stress, Aβ levels, and apoptosis, promotes dendritic growth
and mitochondrial health, improve mood and memory [21]. Crocus sativus (Saffron), a member
of the Iridaceae family has long been known to possess pharmacological properties [37] like
antimicrobial, antioxidant [38], cytotoxic, neuroprotective [39] and antidepressant [40]
properties.
Computational biology approaches serve as invaluable resources in screening potential
therapeutic agents for a particular disease target. Interactions between bioactive
compounds found in the neuroprotective plants and key enzymes involved in Alzheimer’s
disease pathology can be simulated and predicted virtually. Molecular docking [41]
is an in-silico technique used to predict the preferred orientation and conformation
of a small molecule (ligand) when bound to a target protein (receptor). By performing
molecular docking studies, the interaction of natural bioactive compounds with key
enzymes involved in Alzheimer’s disease pathology, such as beta-amyloid protein and
beta-secretase (BACE1) could be understood. Molecular dynamics (MD) [42] is a computational
method used to simulate the movement and behavior of atoms and molecules over time. These
simulations provide insights into the dynamic nature of protein-ligand interactions at
the atomic level. By integrating ADMET prediction and drug-likeness assessment into the
drug discovery process, we can prioritize compounds with favorable pharmacokinetic and
pharmacodynamic properties while minimizing the risk of toxicity and optimizing the
likelihood of clinical success. This investigation aims to bridge the gap between
traditional herbal knowledge and modern computational techniques by exploring the
potential of natural bioactive compounds as novel candidates for Alzheimer’s
disease therapy.
Methods
Retrieval of ligands
The plants selected for the present study Withania somnifera, Bacopa monnieri, Centella asiatica, and Crocus sativus were chosen based on their traditional use as neuroprotective agents and documents efficacy in treating neurological disorders. An extensive literature review guided the selection of 39 phytochemicals, focusing on phytocompounds with diverse chemical classes, known pharmacological activities, and drug-like properties. The drug Donepezil [43] was chosen as a standard control. The 3D-structures of selected ligands and drugs were retrieved from PubChem [44] database (https://pubchem.ncbi.nlm.nih.gov/) in sdf file format. The compound structures were energy minimized using Chem-3D before docking studies. (Table 1)
Table 1.
Major bioactive compounds from selected medicinal plants used for molecular docking.
| Medicinal plants | Common name | Family | Compounds | Compound ID |
|---|---|---|---|---|
| Withania somnifera | Ashwagandha | Solanaceae | Withanone | 21679027 |
| Somniferine | 14106343 | |||
| Withasomnine | 442877 | |||
| (-)-Anaferine | 443143 | |||
| Withaoxylactone | 101687981 | |||
| Withasomniferol A | 101710595 | |||
| Viscosalactone B | 57403080 | |||
| Withasomnilide | 102066413 | |||
| Physagulin-d | 10100412 | |||
| Bacopa monnieri | Brahmi | Scrophulariaceae | Bacoside A | 92043183 |
| Bacopasaponin A | 101995276 | |||
| Plantainoside D | 9986606 | |||
| Cucurbitacin A | 5281315 | |||
| Rosavin | 9823887 | |||
| Bacosterol-3-O-β-D-glucopyranoside | 163184359 | |||
| Bacosine | 71312547 | |||
| Loliolide | 100332 | |||
| Bacopaside I | 21599442 | |||
| Betulinic acid | 64971 | |||
| Centella asiatica | Gotu Kola | Apiaceae | Asiatic acid | 119034 |
| Madecassic acid | 73412 | |||
| Cianidanol | 9064 | |||
| Terminolic acid | 12314613 | |||
| Crocus sativus | Saffron | Iridaceae | Crocetin | 5281232 |
| Safranal | 61041 | |||
| Picrocrocin | 130796 | |||
| Kaempferol 3-sophoroside-7-glucoside | 12960460 | |||
| Crocetin dimethyl ester | 5316132 | |||
| Phytoene | 5280784 | |||
| Zeaxanthin | 5280899 | |||
| Beta-Carotene | 5280489 | |||
| Lycopene | 446925 | |||
| Phytofluene | 6436722 | |||
| Zeta-Carotene | 5280788 | |||
| Caffeic acid | 689043 | |||
| Gallic acid | 370 | |||
| Luteolin | 5280445 | |||
| Chlorogenic acid | 1794427 | |||
| Retinoic acid | 444795 | |||
| Drug | ||||
| Donepezil | 3152 | |||
Retrieval of target proteins and preparation
This investigation targets two major proteins related to Alzheimer’s disease, such as acetylcholinesterase (AChE) (PDB ID: 1B41) and β-secretase (BACE-1) (PDB ID: 1TQF). The 3D structure of selected proteins were downloaded from RCSB Protein Data Bank (https://www.rcsb.org/pdb/) [45] in pdb format (Figure 1). The enzyme AChE is responsible for hydrolysis of acetylcholine to acetic acid and choline. Acetylcholine is a crucial neurotransmitter in the central nervous system playing a significant role in brain signaling and cognitive functioning. A study in the 1970s revealed that patients of Alzheimer’s disease show a deficiency of Acetylcholine in the brain [46]. This leads to a cholinergic deficit, an inability to transmit neurological impulses across cholinergic synapses, and a consequent cognitive decline [47]. AChE has additionally been implicated in the formation of plaque. AChE inhibitors serve as the most effective symptomatic treatment for AD by slowing down the biochemical breakdown of acetylcholine, leading to prolonged neurotransmission. It may also modify the deposition of β-amyloid [48].
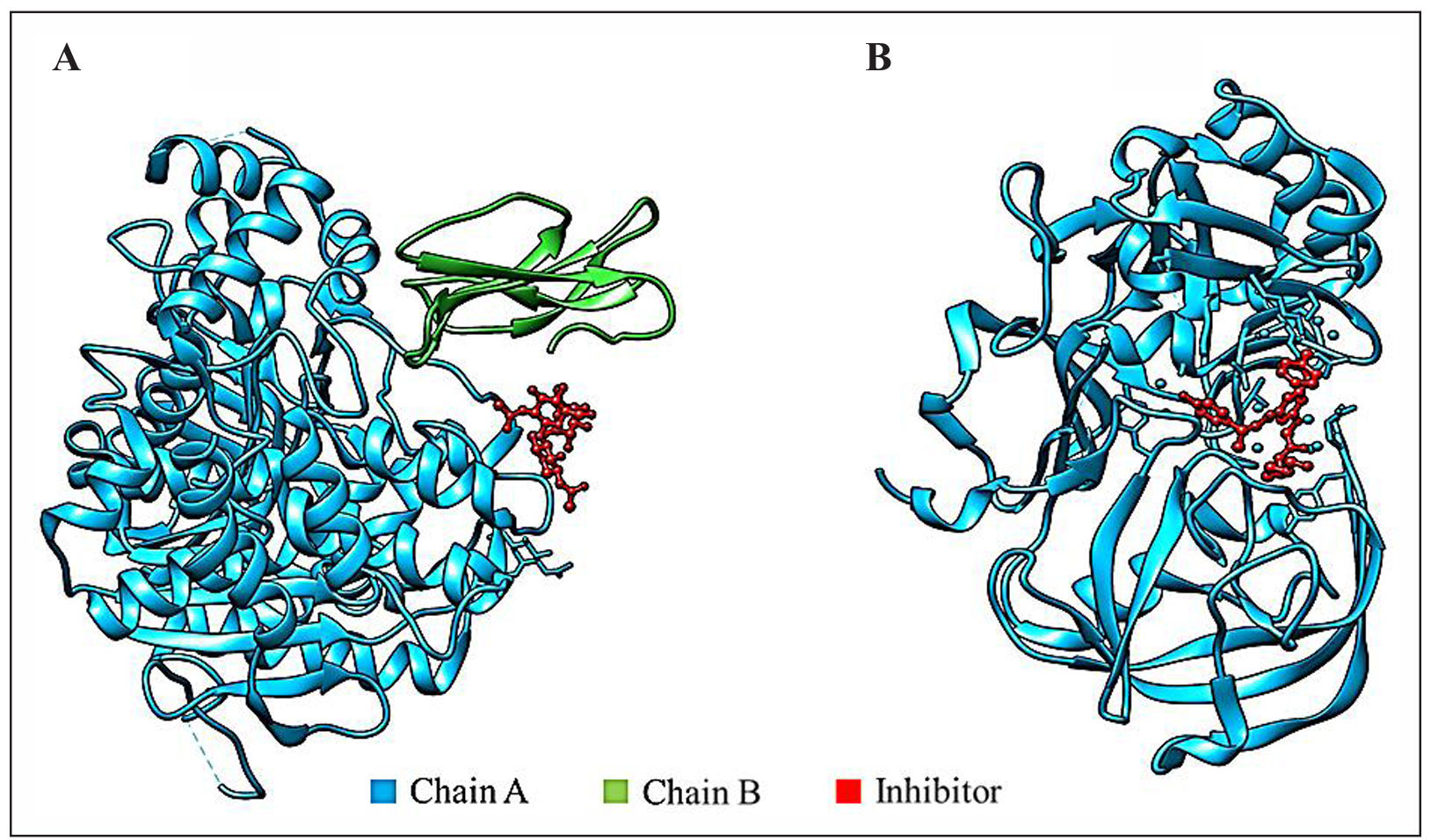
Figure 1. 3D Structures of target proteins. (A) Acetylcholinesterase enzyme. (B) β-secretase enzyme. Both proteins show the presence of inhibitors in the structure
BACE-1 enzyme is accountable for the aberrant degradation of APP [10], which
play a role in neuronal growth and repair. The BACE-1 cleaves APP, resulting
in the production of insoluble peptides that aggregate to form beta-amyloid plaques.
The present investigation aims to examine the potential inhibitory effects of diverse
bioactive compounds on the BACE-1 enzyme. By specifically targeting BACE-1, the abnormal
degradation of amyloid precursor protein (APP) and subsequent formation of plaque can
be mitigated.
Both the proteins were prepared before docking by the removal of water molecules,
heteroatoms, and extra ligands. Then checking and repairing missing atoms, the addition
of polar hydrogens, the addition of Kollman charges, and spreading the charge deficit
equally. Grid maps were also created for the proteins covering their active sites.
For 1B41, the grid size was set to 74×74×74 (XYZ) points, and the coordinates for the
grid center were set to x= 126.361, y= 102.556, and z= -121.778. For 1TQF, the grid
size was set at 50×50×50 (XYZ) and the grid-centre coordinates were x= 28.250, y= 45.805,
and z= 2.86.
Molecular docking
Molecular docking between ligands and target proteins is carried out to analyze the fit of ligands in protein’s active site [41]. The binding energy of each ligand is calculated for different poses/conformations of the ligand with the protein. The ligands and conformations showing the least binding energy indicate a favorable and stable fit with the protein. Autodock vina software [49] was used to perform the molecular docking between selected ligands and target proteins. Discovery studio (DS) was used to assess the optimal configuration for each ligand-protein interaction according to binding energies. The ligands with the highest binding energy in the interaction profile were chosen for further examination through molecular dynamics simulations.
Molecular dynamics
The present investigation employed molecular dynamics (MD) simulations using the academic version of the Desmond program (version 2.0) to assess the structural stability of receptor-ligand complexes) [50]. The system in this program was constructed using the TIP3P water model [51] with a cubic periodic box containing simple point charge (SPC) (10Å×10Å×10Å) and optimised potentials for liquid simulations (OPLS) all-atom force field 2005 [52]. The system was subsequently neutralized by introducing the requisite amount of sodium ions. A receptor-ligand complex was provided for the energy minimization step and pre-equilibration for various confined steps. The molecular dynamic simulations were conducted using the OPLS 2005 force field parameters and periodic boundary conditions in the NPT ensemble system [53, 54]. The system was maintained at a constant temperature of 300 K with a relaxation time of 1 ps. The volume was kept constant, and the smooth particle mesh ewald (PME) method was employed with a tolerance limit of 10-9 mm. A cut-off distance of 9.0Å was used. The investigation of protein structures was conducted at a production time of 100 ns, with evaluations performed every 1 ns. To determine the stability, a model structure from the molecular dynamics (MD) simulation during the production phase was chosen. In addition, the root mean square deviation (RMSD) and root mean square fluctuation (RMSF) to examine any structural changes that occurred during the dynamic interaction between the receptor and ligand complexes were also investigated [55].
Binding free energy calculations
The binding free energies of protein-ligand complexes have been calculated using MM-GBSA and molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) [56, 57]. As a result, the PRIME module of Maestro 11.4 and the OPLS-2005 force field were employed to calculate the binding energy of the best-docked ligand-receptor complex using the equation below:∆GBind = ∆EMM + ∆GSolv + ∆GSA
where ∆EMM is the difference of the minimized energies of the protein-ligand complex, while ∆GSolv is the difference between the GBSA solvation energy of the protein-ligand complexes and the sum of the solvation energies for the protein and ligand. ∆GSA indicates the surface area energies in the protein-ligand complexes and the difference in the surface area energies for the complexes [58].
ADME and toxicity analysis
Various characteristics were examined to evaluate the drug-like properties of the selected phytochemicals. Lipinski’s rule, executed via molinspiration, was employed to analyze characteristics like the number of hydrogen acceptors (less than 10), the number of hydrogen donors (less than 5), molecular weight (more than 500 Daltons), and partition coefficient log P (not less than 5) [59]. The selected phytocompounds were subjected to their toxicity attributes. The 3D structures of the best-docked phytocompounds and Donepezil were converted to SMILES format and evaluated for toxicity using admetSAR [60] and PROTOX-III webserver [61].
Results
Molecular docking
The results with the least (most negative) binding energy values are considered strong interactions and reflect a stable and favorable fit. The binding energies (kcal/mol) of the selected compounds with the two target proteins (1B41 and 1TQF) are tabulated in Table 2. Retinoic acid showed a strong binding energy of -9.2 kcal/mol with 1B41 protein, while Somniferine showed the highest binding energy value of -8.8 kcal/mol with 1TQF protein. Donepezil showed binding energy values of -8.3 kcal/mol and -8.4 kcal/mol with 1B41 and 1TQF, respectively (Table 2).
Table 2.
Binding energy (kcal/mol) of selected phytocompounds and drugs with target proteins.
| S. No | Phytocompounds | Free binding energy (kcal/mol) | |
|---|---|---|---|
| 1B41 | 1TQF | ||
| 1 | Withanone | -8.7 | -7.4 |
| 2 | Somniferine | -8.8 | -8.8 |
| 3 | Withasomnine | -8.0 | -6.8 |
| 4 | (-)-Anaferine | -7.0 | -6.9 |
| 5 | Withaoxylactone | -8.9 | -7.7 |
| 6 | Withasomniferol A | -8.2 | -8.7 |
| 6 | Viscosalactone B | -8.6 | -7.1 |
| 7 | Physagulin-d | -8.6 | -8.7 |
| 8 | Physagulin-d | -8.6 | -8.7 |
| 9 | Bacopasaponin A | -8.4 | -7.1 |
| 10 | Plantainoside D | -8.8 | -8.5 |
| 11 | Cucurbitacin A | -8.2 | -7.6 |
| 12 | Rosavin | -8.4 | -7.6 |
| 13 | Bacosterol-3-O-β-D-glucopyranoside | -8.5 | -7.2 |
| 14 | Bacosine | -7.7 | -6.7 |
| 15 | Loliolide | -6.8 | -5.6 |
| 16 | Bacopaside I | -8.7 | -7.7 |
| 17 | Asiatic acid | -7.6 | -6.2 |
| 18 | Madecassic acid | -8.1 | -7.1 |
| 19 | Cianidanol | -7.9 | -7.7 |
| 20 | Terminolic acid | -7.7 | -7.4 |
| 21 | Crocetin | -7.6 | -7.4 |
| 22 | Safranal | -6.8 | -5.4 |
| 23 | Picrocrocin | -7.8 | -6.1 |
| 24 | Betulinic acid | -8.0 | -7.8 |
| 25 | Caffeic acid | -6.8 | -5.6 |
| 26 | Gallic acid | -6.8 | -5.5 |
| 27 | Chlorogenic acid | -8.7 | -8.1 |
| 28 | Luteolin | -8.6 | -7.5 |
| 29 | Retinoic acid | -9.2 | -7.6 |
| 30 | Donepezil (Drug) | -8.3 | -8.4 |
Discovery Studio Visualizer was used to analyze the interactions of retinoic acid with 1B41, and Somniferine with 1TQF (Figure 2). Retinoic acid formed a hydrogen bond with HIS (A):287, and two pi-sigma bonds with TYR (A):341 and TRP (A):286 residues of 1B41 protein. Other non-covalent interactions were formed with TYR (A):124 and VAL (A):294 residues of 1B41. Somniferine was found to form two hydrogen bonds with LYS (A):321 and ARG (A):307 residues of 1TQF protein. Other non-covalent interactions were formed with ILE (A):110 and ARG (A):307 residues of 1TQF. Interactions of Donepezil with both the target proteins are also shown in Table 3.
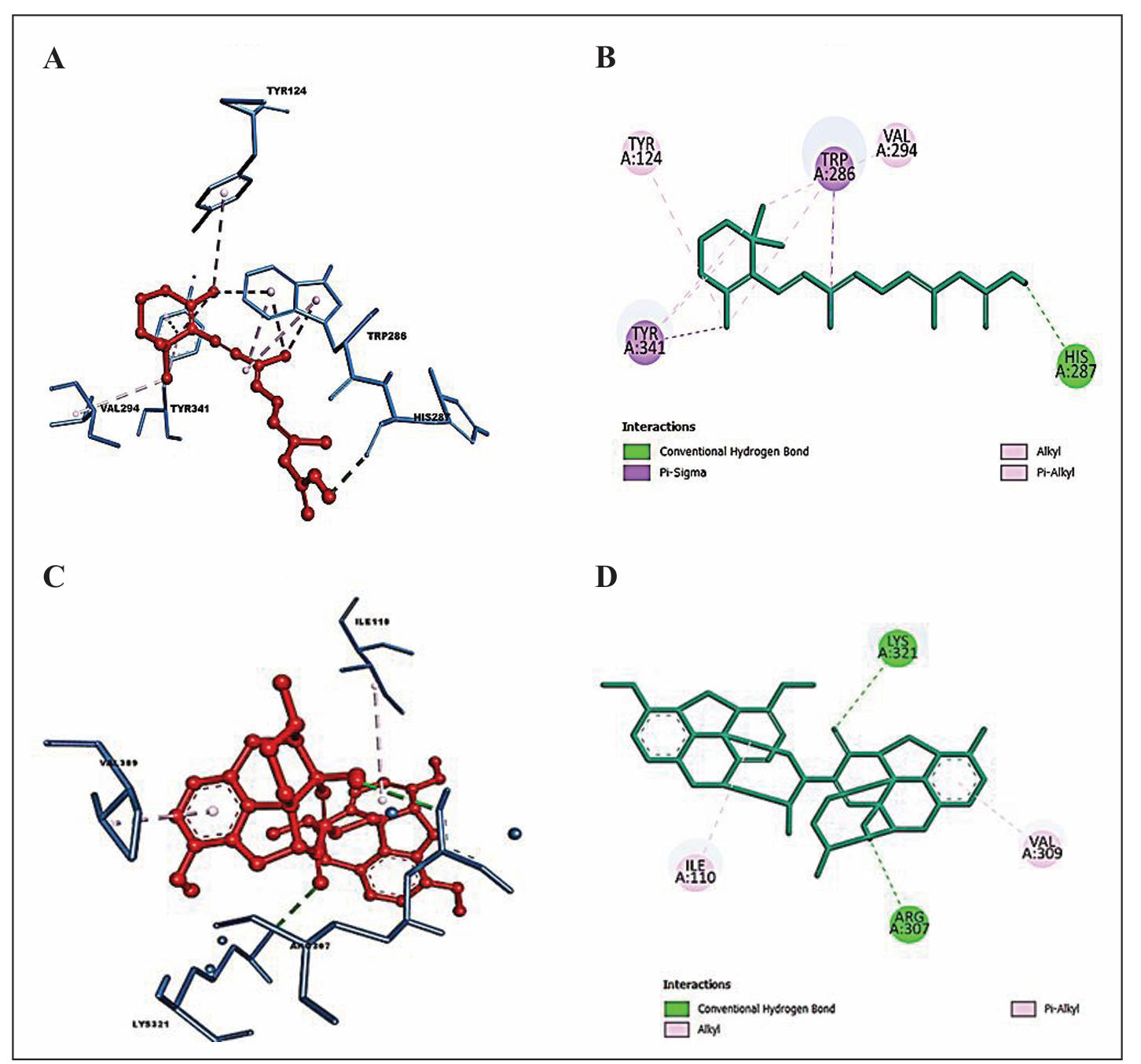
Figure 2. Docked pose of the top-ranked ligands with target proteins. (A) 3-D interactions of retinoic acid with interacting amino acids of 1B41. (B) 2-D interactions of retinoic acid with interacting amino acids of 1B41. (C) 3-D interactions of Somniferine with interacting amino acids of 1TQF. (D) 2-D interactions of Somniferine with interacting amino acids of 1TQF.
Table 3.
Binding interactions of Retinoic acid, Somniferine, and Donepezil with target proteins.
| Protein | Ligand | No. of hydrogen bonds | Hydrogen bonds | Non-covalent interactions |
|---|---|---|---|---|
| 1B41 | Retinoic acid | 1 | His (A):287) | Tyr (A):341, Trp (A):286, Tyr (A):124, Val (A):294 |
| Donepezil | 2 | Tyr (A):72, Ser (A):293 | Tyr (A):341, Trp (A);286, Tyr (A):124, Phe (A):2997, Leu (A):289 | |
| 1TQF | Somniferine | 2 | Lys (A):321, Arg (A):307 | Ile (A:110), Arg (A):307 |
| Donepezil | 6 | Arg (A):307, Lys (A):321, Gly (A):230, Asp (A):32, Thr (A):72, Tyr (A):198 | Val (A):309, Tyr (A):71, Pro (A):70 |
Molecular dynamics
Molecular dynamics simulation is used to study the dynamic behavior of protein-ligand complexes over time. It provides insights into the motions, interactions, and structural changes of atoms and molecules at the atomic or molecular level within a protein-ligand complex. Retinoic acid and Somniferine demonstrated strong binding energy when interacting with 1B41 and 1TQF, respectively. Subsequently, the protein-ligand complexes of retinoic acid-1B41 and somniferine-1TQF were screened through molecular dynamics simulations for 100 ns.
RMSD analysis of protein-ligand complexes
The RMSD reflects the average deviation in the positions of selected atoms of a simulated complex from a reference structure over a trajectory. The deviation for each frame in the trajectory is calculated. Figure 3 displays the RMSD evolution of the protein (left y-axis) and the ligand (right y-axis). The starting point is defined by the docked arrangement of the ligand and the protein in the complex. The deviation from this reference position during the MD simulation is then gauged by aligning all protein-ligand frames obtained throughout the trajectories. The term "lig fit prot" (line colored in magenta) implies the RMSD of the ligand from the backbone of the protein. For retinoic acid-1B41 complex, the protein backbone hovers within the range of 2Å-3.4 Å and fluctuation initially up to 30 ns. The complex was then stable between 30 to 80 ns in between 2-2.8 Å, then showed fluctuation between 80-90 ns between 2.0-3.2 Å (Figure 3A). The RMSD plot of somniferine-1TQF complex (Figure 3B) shows a more fluctuating curve. The protein backbone hovers in the range from 1.2 Å to 2.8 Å from 0-100 ns. The lig-fit-prot curve remained well below the protein backbone curve in both protein-ligand complexes, indicating that the orientation of the ligand remains the same (Figure 3).
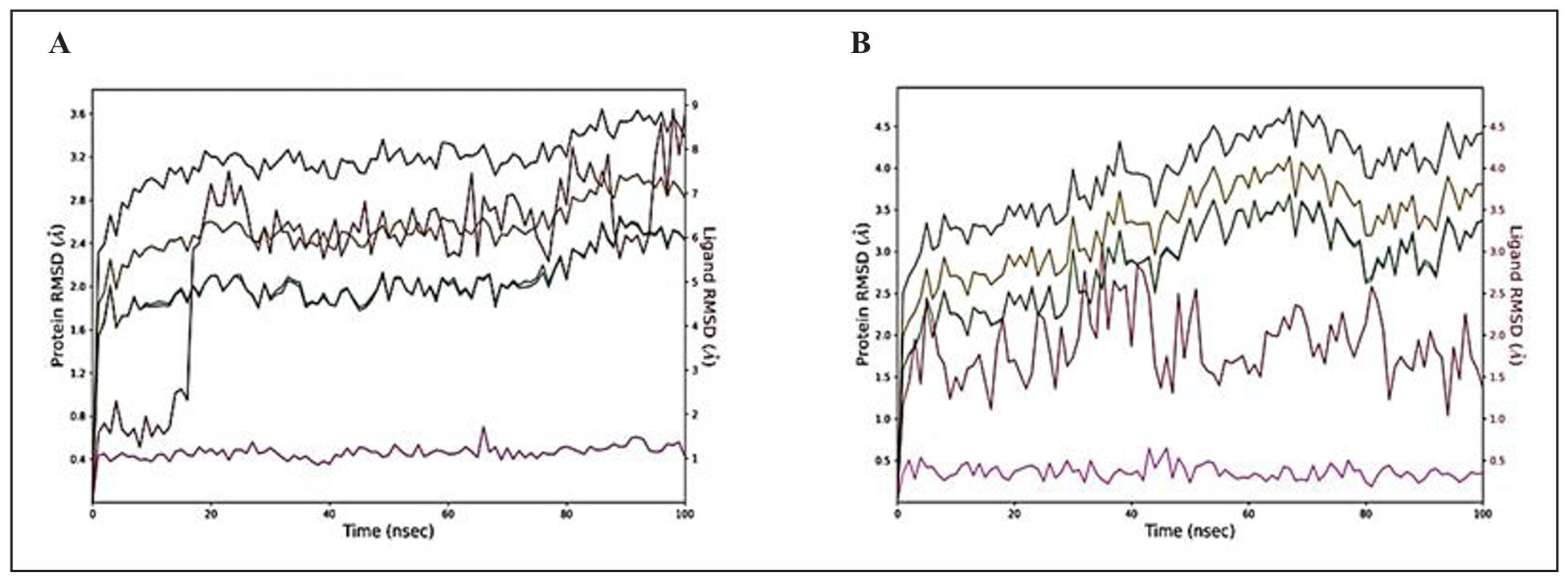
Figure 3. RMSD graphs of protein-ligand complexes for 100 ns. (A) Retinoic acid complexed with 1B41. (B) Somniferine complexed with 1TQF protein. The following are the color legends: heavy ions (yellow), Cα (blue), side chains (green), ligand with protein (dark pink), and ligand with ligand (pink).
RMSF analysis of protein-ligand complexes
The RMSF measures the localized structural changes along the protein chain during
molecular dynamics simulations. Peaks on the RMSF plot indicate regions of the
protein that experience the maximum fluctuations throughout the simulation.
Secondary structure elements, such as α-helices and β-strands, highlighted with
red and blue backgrounds, respectively, are characterized by lower fluctuations,
signifying their inherent rigidity. In contrast, loop regions, often unstructured,
tend to exhibit higher fluctuations, emphasizing their dynamic nature. The
green-colored vertical bars mark the protein residues that encounter the ligand.
These interactions play a crucial role in understanding the binding dynamics and
stability of the complex.
To complement the RMSF analysis, the plot also correlates the fluctuation data
with experimental B-factors. While the RMSF and B-factor definitions differ,
a parallel between simulation results and crystallographic data is anticipated.
This correlation enhances the reliability of the MD simulation outcomes, offering
a valuable perspective on the agreement between computational predictions and
experimental observations. The trajectory of 1B41-retinoic acid complex shows
a plot with minimal residual fluctuations in the range of 0.5-1.2 Å and almost
parallel RMSF and B-Factor curves (Figure 4A). The plots of Somniferine with
1TQF initially displayed few differences in the RMSF and B-Factor values (Figure 4B)
till residue index 100-120, and after that they show overlapping RMSF and B-Factor
curves. 1TQF-somniferine displayed residual fluctuation in the range of 1.0-2.0 Å.
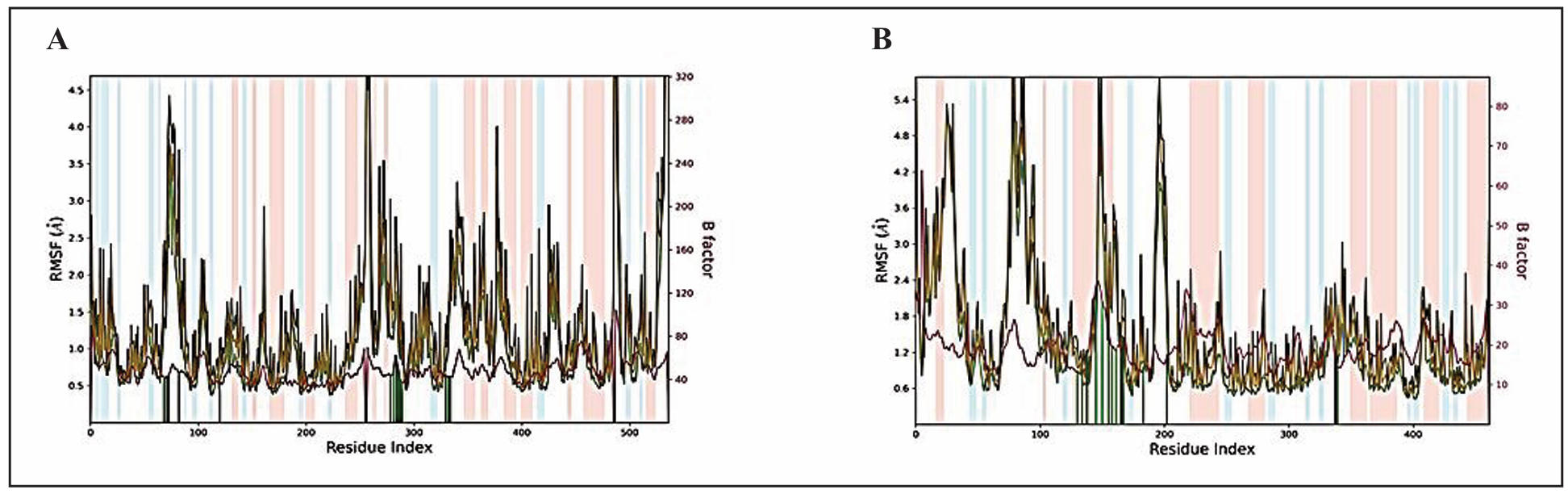
Figure 4. RMSF plot of the protein-ligand complexes for 100 ns. (A) Retinoic acid complexed with 1B41. (B) Somniferine complexed with 1TQF protein. The color legends are as follows: Cα (blue), backbone (green), heavy atoms (brown), and B factor (dark pink).
Ligand properties
In the MD simulation, the behaviour of the two best performing phytocompounds, Retinoic acid and Somniferine was assessed over a 100 ns timeframe, with key properties evaluated to understand their stability and interaction with the surrounding environment (depicted in Figure 5). For the retinoic acid, The Root Mean Square Deviation (RMSD) fluctuated between 0.5 Å and 1.5 Å while for Somniferine, the range remained below 0.6 Å, indicating that the ligands maintained a stable structure with minor deviations from their initial conformations (time t = 0). The Radius of Gyration (rGyr), which measures the ‘extendedness’ of the ligand, fluctuated slightly between 4.8 Å to 5.1 Å for Retinoic acid and between 5.12 Å and 5.28 Å for Somniferine, showing that the ligands retained compact shape throughout the simulation. Notably, no intramolecular hydrogen bonds (intraHB) were detected in both simulations, suggesting that the ligands did not rely on internal hydrogen bonds to maintain their structural stability. The molecular surface area (MolSA), which represents the molecular surface calculated with a 1.4 Å probe radius (comparable to the van der Waals surface), ranged from 332.5 Ų to 340 Ų in Retinoic acid and between 474 Ų - 486 Ų in Somniferine, indicating minimal changes in the ligand’s surface. The Solvent Accessible Surface Area (SASA), which measures the extent to which the ligand is exposed to solvent (water), fluctuated between 50 Ų and 200 Ų for Retinoic acid and between 120 Ų and 300 Ų in Somniferine. Higher SASA values could imply that the ligand is more exposed and possibly at risk of being pulled away from the active site of the protein by the solvent, while lower values indicate stronger interaction with the protein. The polar surface area (PSA), which accounts for the solvent-accessible areas contributed by polar atoms (oxygen and nitrogen), ranged from 87 Ų to 93 Ų in retinoic acid and 150 Ų to 168 Ų in somniferine.
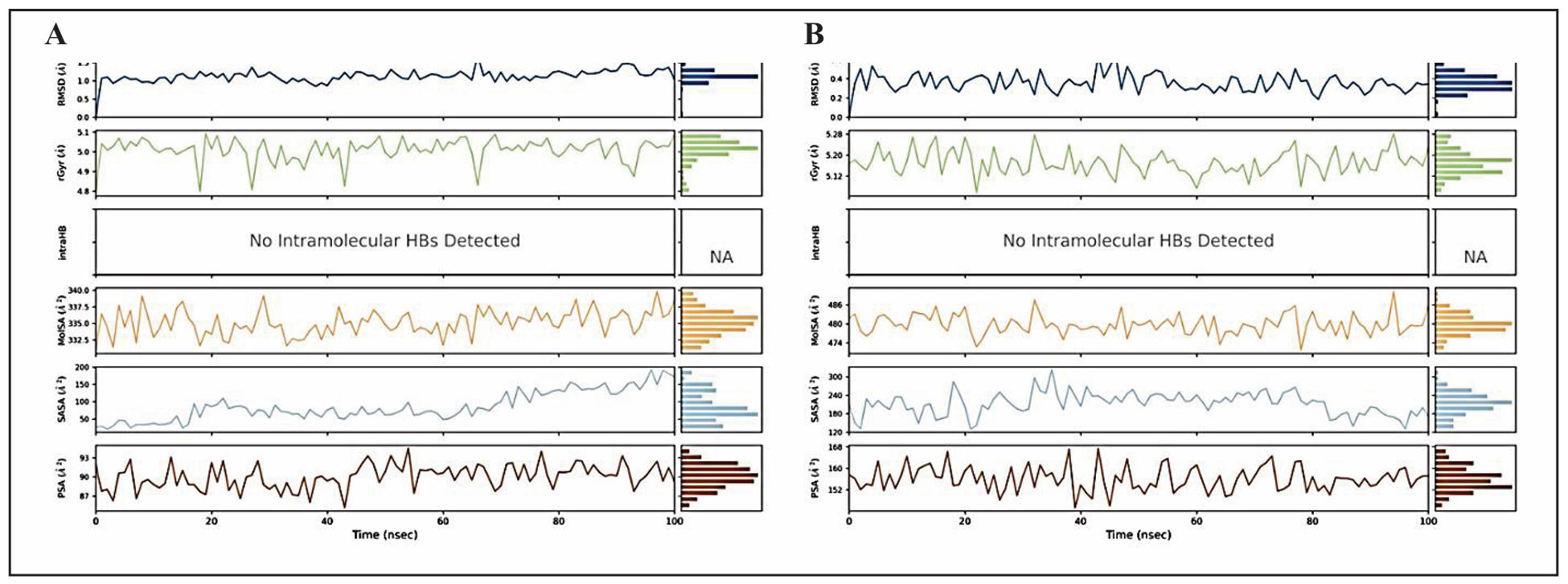
Figure 5. Ligand properties for best-docked phytocompounds. (A) Retinoic acid on interacting with 1B41 protein and (B) Somniferine on interacting with 1TQF protein during MD simulation such as RMSD, the radius of gyration (rGyr), intramolecular hydrogen bonds (intraHB), Molecular Surface Area (MolSA), Solvent Accessible Surface Area (SASA), Polar Surface Area (PSA).
Protein-ligand contacts histogram
The interactions between the protein and the ligand are studied throughout the simulation and depicted through the histogram. Four types of interactions are summarized in the plots (Figure 6): hydrogen bonds (green), hydrophobic (grey), ionic (pink), and water bridges (blue). Retinoic acid complexed with 1B41 showed one ionic bond with Asp (A): 74; water bridges with Asn (A): 283, His (A): 287, Leu (A): 289, Pro (A): 290, and Arg (A): 296; polar contacts with Leu (A): 76, Tyr (A): 77, Trp (A): 86, Tyr (A): 124, Phe (A): 295, Phe (A): 297, Tyr (A): 337, Phe (A): 338 and Tyr (A): 341 (Figure 6A). Somniferine formed the polar contacts interactions with Phe (A):148, Met (A):149, Arg(A):156, Phe (A):161, Leu (A):166, Pro 178, Met (A):194, Phe (A): 213 residues of 1TQF protein (Figure 6B). Figure 6C and 6D depict the interactions of crucial amino acids of selected target proteins with retinoic acid and Somniferine. Several residues establish several explicit interactions with the ligand, as seen by a more diffuse shade of orange, as indicated by the scale on one side of the figure. These plots are extremely important because they show that retinoid acid and Somniferine interact with the amino acids of target proteins throughout the simulation and do not dissociate from their interacting site. On the other hand, small changes in the ligand’s RMSD and RMSF values, as shown in Figure 2 and 3, respectively, imply that the ligands may be reorienting themselves during the simulation.
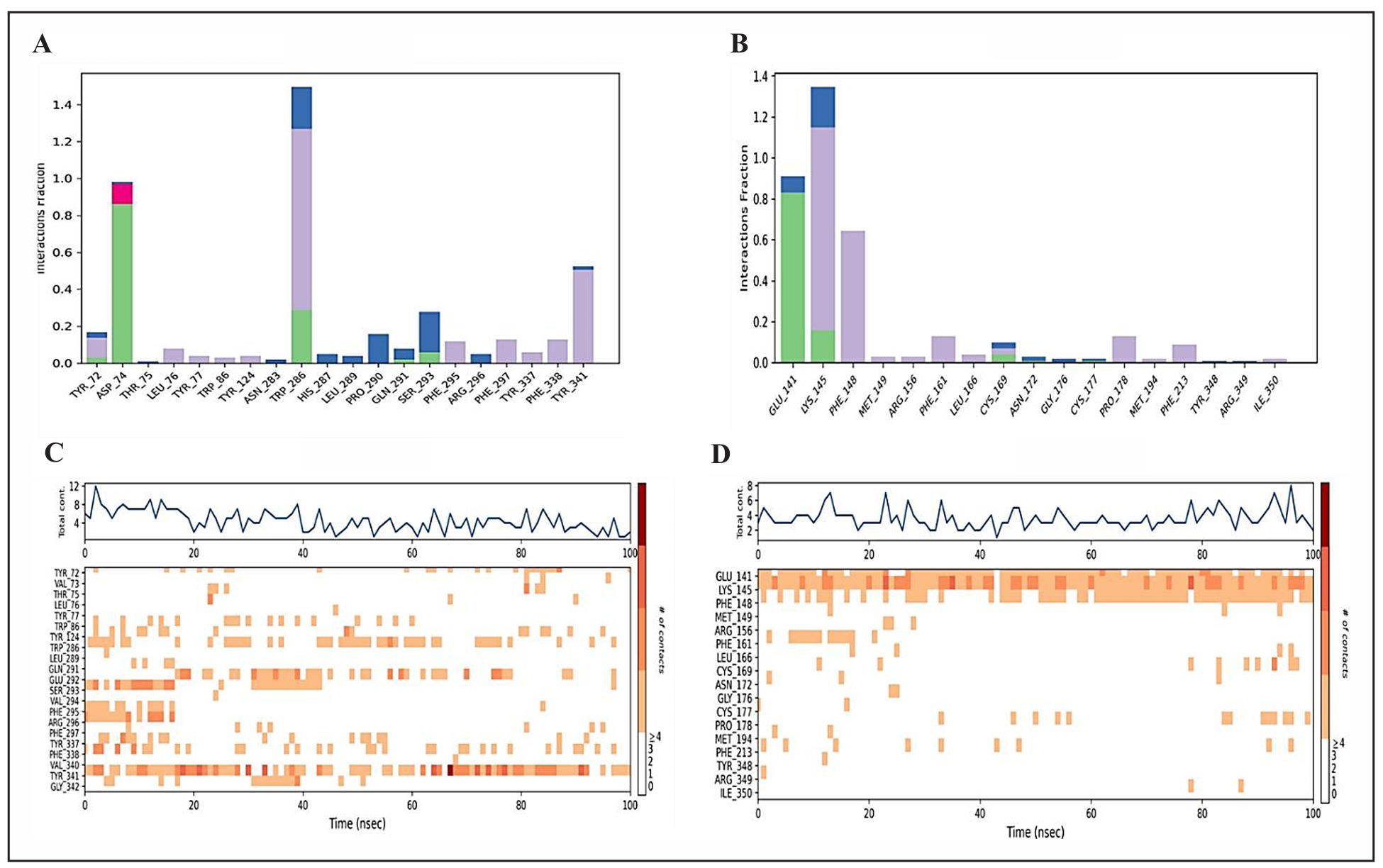
Figure 6. Interaction profiles of protein-ligand complexes. (A) Interaction profile of retinoic acid-1B41 protein. (B) Interaction profile of Somniferine-1TQF protein. (C) Timeline representation of the interactions of amino acids of 1B41 protein with retinoic acid, (D) timeline representation of the interactions of amino acids of 1TQF protein with Somniferine. The bars are colored to show different types of interactions, such as hydrogen bonds (green), polar contacts (purple), and water-bridges (blue).
Preservation of intermolecular contacts in molecular dynamics simulations
In the 1B41-Retinoic acid complex, the ligand interacts with the TYR341 residue of the protein. The interaction is primarily hydrophobic, indicated by the green label, reflecting non-polar side chain interactions between the tyrosine residue and the hydrophobic components of the ligand. The ligand’s structure, which includes extensive conjugation represented by alternating single and double bonds in the E-configuration along with bulky cyclic structures, enhances this interaction by maximizing hydrophobic contact. In the 1TQF-Somniferine complex, several key interactions were observed during the molecular dynamics simulation. The GLU (A):141 residue forms a hydrogen bond with the ligand, preserved for 83% of the simulation time, and contributes to stabilizing the interaction through electrostatic attraction due to its negative charge. The PHE (A):148 residue engages in a Pi-Pi stacking interaction with the ligand’s aromatic rings, with a 41% occurrence during the simulation, enhancing hydrophobic interactions. Additionally, the LYS (A):145 residue forms a Pi-cation interaction with the ligand, maintained for 99% of the simulation time, reflecting a highly stable contact between the positively charged lysine residue and the ligand’s aromatic ring. These interactions, including hydrogen bonds, Pi-Pi stacking, and Pi-cation contacts, play a significant role in maintaining the structural stability of the ligand-protein complex. Figure 7 presents the 2D interaction maps of the best-docked compounds, illustrating the maintenance of contacts throughout the simulation trajectory.
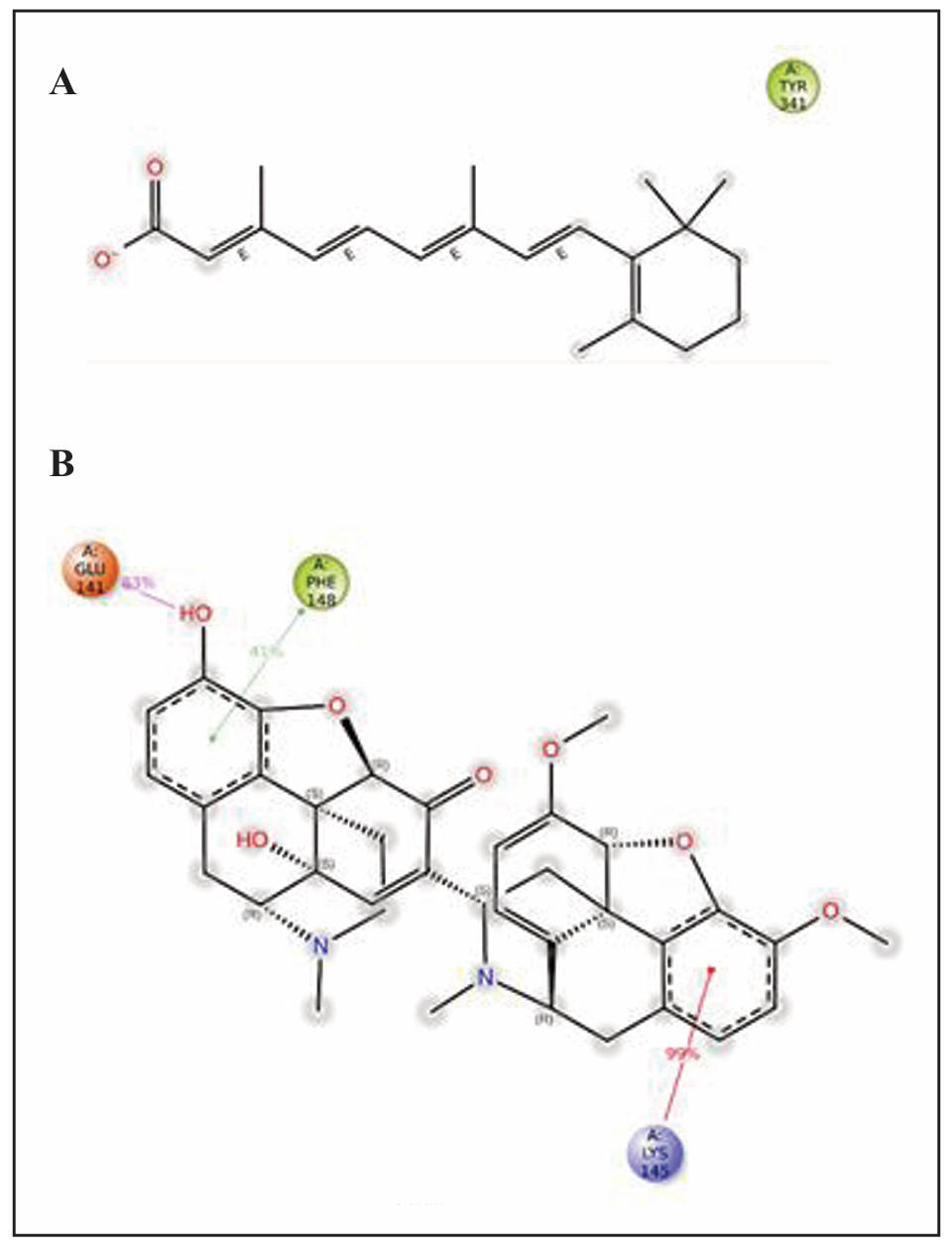
Figure 7. Preserved contacts of Retinoic acid with 1B41 (A) and Somniferine with 1TQF (B) proteins, captured during MD simulations.
Binding-free energy calculations
Post-simulation analysis of both protein-ligand complexes was carried out by capturing snapshots of the trajectory profiles generated during MD simulations, as shown in Table 4. It was observed that both protein-ligand complexes exhibited negative ∆Gbinding energies, suggesting the stability of these complexes during the MD simulation. The results showed that the Van der Waals interactions (∆GvdW) were -44.13 ± 5.22 kcal/mol for the retinoic acid-1B41 complex and -56.97 ± 2.76 kcal/mol for Somniferine-1TQF complexes, indicating that both the ligands remain close to the interacting amino acids of the target proteins. The Coulomb energy displayed a negative value in all complexes, indicating consistently low potential energy for ligands when bound to their respective target proteins. The finding indicates that protein-ligand complexes generally exhibit improved stability. Table 4 provides an extensive overview of the contributions made by different components, such as hydrogen bonding and covalent bonding, to the total energy, along with the associated total energy values.
Table 4.
MM/GBSA profiles of best-docked phytocompounds during interaction with target proteins
| Proteins | Ligands | ∆GBind (kcal/mol) |
∆GCoulomb (kcal/mol) |
∆GvdW (kcal/mol) |
∆GH-bond (kcal/mol) |
∆GCovalent (kcal/mol) |
|---|---|---|---|---|---|---|
| 1B41 | Retinoic acid | -39.45 ± 6.94 | -32.53 ± 12.08 | -44.13 ± 5.22 | -0.54 ± 0.05 | 2.22 ± 1.16 |
| 1TQF | Somniferine | -68.13 ± 4.43 | -17.11 ± 3.21 | -56.97 ± 2.76 | -0.49 ± 0.13 | 2.16 ± 0.77 |
Drug-likeness and toxicity analysis
Table 5 displays the results of Lipinski’s rule of five for the best-docked phytocompounds and selected drugs that were chosen. Both the selected phytocompounds and selected drugs were found to follow Lipinski’s rule of five. The results from toxicity prediction showed that retinoic acid and Somniferine were non-neurotoxic, non-carcinogenic, and non-cytotoxic in nature, whereas, the drug, Donepezil was positive for all these parameters. Retinoic acid was positive for hepatotoxicity, while Somniferine was immunogenic in nature. The predicted LD50 (mg/kg) was found to be 1100 (Class-4) for both retinoic acid and Somniferine, whereas the predicted LD50 for Donepezil was found to be 550 mg/kg (Class-4), indicating the non-toxicity of both the phytocompounds.
Table 5.
Binding energy (kcal/mol) of selected phytocompounds and drug with target proteins.
| Type of Parameter | Parameters | Ligands | Drug | |
|---|---|---|---|---|
| Retinoic acid | Somniferine | Donepezil | ||
| Drug-likeness | mLogP (< 4.15) | 5.01 | 2.74 | 4 |
| nrot (< 5) | 5 | 3 | 4 | |
| MW (< 500Da) | 300.44 | 608.68 | 379.49 | |
| HBD (< 5) | 1 | 2 | 0 | |
| HBA (< 10) | 2 | 9 | 4 | |
| Lipinski rule | Yes | Yes | Yes | |
| Toxicity Prediction | Hepato- | Yes | No | No |
| Neuro-toxicity | No | No | Yes | |
| Carcinogenicity | No | No | Yes | |
| Cyto-toxicity | No | No | Yes | |
| Immunogenicity | No | Yes | Yes | |
| LD50 (mg/kg) | 1100 mg/kg Class: IV | 1100 mg/kg Class: IV | 505 mg/kg Class: IV | |
Note: MlogP, measure of molecular hydrophobicity; nrot, number of rotatable bonds; MW, molecular weight; HBA, H-bond acceptor; HBD, H-bond donor; LD50, Lethal dose.
Discussion
Computational drug discovery methods have emerged as efficient pillars
in the lifecycle of drug-development [62]. Taking the ‘experimental set-up’ to ‘virtual
environment’ has allowed the screening of even billions of molecules for their
drug-likeness properties against many disease targets [63]. In-silico analyses
have significantly reduced the time and cost of identifying suitable molecules
as drugs, expected to reduce clinical trial failures at later stages. The chemical
space of compounds that can be used as drugs is huge, with a large part of them
coming from traditional herbal knowledge. Natural compounds have shown promising
therapeutic potential and reduced toxicity values [64] and provide cost-effective
measures in terms of synthesis and collection of raw material. Building upon
traditional knowledge of such natural herbs, this investigation aimed to find
potential therapeutic compounds to treat Alzheimer’s disease.
Several plants have long been used traditionally as brain tonics and neuroprotective
agents [65]. Our study aimed to explore the binding interactions between natural
compounds from such neuroprotective herbs including Withania somnifera, Bacopa
monnieri, Centella asiatica, and Crocus sativus, and enzymes associated with
Alzheimer’s disease. Among the 39 compounds screened, retinoic acid
(PubChem ID: 444795) and somniferine (PubChem ID: 14106343) emerged as promising
candidates for Alzheimer’s disease therapy as they exhibited strong binding
affinities (-9.2 kcal/mol and -8.8 kcal/mol, respectively) and stable interactions
with AChE (PDB ID: 1B41) and BACE-1 (PDB ID: 1TQF), respectively. Prior research
emphasizes the significance of AChE and BACE-1 inhibition in mitigating cognitive
symptoms linked to Alzheimer’s [66-68]. Retinoic acid was established as a potent AChE
inhibitor in this study, a key target for Alzheimer’s treatment [69, 70]. BACE-1
inhibitors are another class of compounds studied against Alzheimer’s [11, 47].
Somniferine has been estimated to be a promising BACE-1 inhibitor in this investigation.
Phytocompounds like Withaferine [71] and Withanolides [72] from Withania somnifera
have been studied by researchers for their potential against AD. Our study established
Somniferine, an alkaloid found in Withania as another novel candidate for Alzheimer’s
disease treatment. Another compound, retinoic acid, found in many plants including
Crocus sativus was established as a lead compound to manage Alzheimer’s disease via
AChE inhibition. Retinoic acid has also been previously studied as a therapeutic
option for AD through cholinergic restoration [73]. Additionally, computational
toxicity prediction plays a crucial role in drug discovery by facilitating early
compound screening and reducing clinical trial failures [74]. The favorable ADMET
properties of retinoic acid and Somniferine underscore their potential as viable
Alzheimer’s treatment candidates. However, in silico analyses are preliminary,
and have their limitations. Understanding how drugs interact with the body and
predicting their real-world outcomes can only be confirmed through experimental
validation, given the dynamic and complex nature of biological systems. It’s
imperative to conduct comprehensive in vitro and in vivo studies to thoroughly assess
the efficacy and toxicity profile of lead compounds.
Conclusions
In conclusion, this research aims to connect traditional herbal wisdom with modern scientific methods, investigating how bioactive compounds from traditional herbs could be used therapeutically for Alzheimer’s disease. The study focused on 39 key compounds found in neuroprotective herbs and employed a cheminformatics approach, combining computational biology techniques such as molecular docking and molecular dynamics simulations with pharmacological and ADMET analyses. The selected compounds were thoroughly analyzed for drug-likeness, pharmacokinetic properties, and toxicity, revealing valuable insights into their potential as drug candidates for Alzheimer’s disease therapy. Among 39 selected phytocompounds, retinoic acid and Somniferine exhibited particularly promising binding interactions, demonstrating adherence to the Lipinski rule, and showing low toxicity. Both the phytocompounds outperformed the standard AD drug, Donepezil, in terms of binding affinity and toxicity properties. The molecular dynamics simulations provided a dynamic perspective on the stability and fluctuations of the protein-ligand complexes over time. Retinoic acid and somniferine displayed stable interactions with respective target enzymes (1B41 and 1TQF). However, further experimental validation, including in vitro and in vivo studies, will be crucial to substantiate the observed interactions and therapeutic potential. This research serves as a promising point for unraveling the hidden pharmacological treasures within nature. By merging traditional knowledge with cutting-edge computational techniques, the study contributes to the ongoing quest for innovative treatments for Alzheimer’s disease, a global healthcare challenge.
Declarations
Authors’ contributions
Conceptualization: Kumar S, Gupta A, and Kumar V; Methodology: Kumar S, Gupta A; Software and Validation: Patel CN, Kumar V; Investigation: Kumar S, Gupta A; Writing—original draft preparation: Kumar S, Gupta A; Writing—review and editing: Kumar V, Kumar A; Visualization: Patel CN, Kumar V; prevision: Kumar V, Kumar A. All authors have read and agreed to the published version of the manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
References
1. (2023). "World Health Organisation. Dementia [cited 2024 Mar 17]." from Available from: https://www.who.int/news-room/fact-sheets/detail/dementia
2. Seeman P, & Seeman N. Alzheimer’s disease: β-amyloid plaque formation in human brain. Synapse, 2011, 65(12): 1289-1297. [Crossref]
3. Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, & Berry RW. Tau, tangles, and Alzheimer’s disease. Biochim Biophys Acta, 2005, 1739(2-3): 216-223. [Crossref]
4. Goedert M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci, 1993, 16(11): 460-465. [Crossref]
5. Nelson PT, Braak H, & Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol, 2009, 68(1): 1-14. [Crossref]
6. Serrano-Pozo A, Frosch MP, Masliah E, & Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med, 2011, 1(1): a006189. [Crossref]
7. Scheff SW, DeKosky ST, & Price DA. Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol Aging, 1990, 11(1): 29-37. [Crossref]
8. Scheff SW, Price DA, Schmitt FA, & Mufson EJ. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging, 2006, 27(10): 1372-1384. [Crossref]
9. Selkoe DJ, & Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med, 2016, 8(6): 595-608. [Crossref]
10. Vassar R. BACE1: the beta-secretase enzyme in Alzheimer’s disease. J Mol Neurosci, 2004, 23(1-2): 105-114. [Crossref]
11. Maia MA, & Sousa E. BACE-1 and γ-secretase as therapeutic targets for Alzheimer’s disease. Pharmaceuticals (Basel), 2019, 12(1): 41-51. [Crossref]
12. Laßek M, Weingarten J, Einsfelder U, Brendel P, Müller U, & Volknandt W. Amyloid precursor proteins are constituents of the presynaptic active zone. J Neurochem, 2013, 127(1): 48-56. [Crossref]
13. Guo T, Zhang D, Zeng Y, Huang TY, Xu H, & Zhao Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol Neurodegener, 2020, 15(1): 40-52. [Crossref]
14. Thal DR, Griffin WS, de Vos RA, & Ghebremedhin E. Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathol, 2008, 115(6): 599-609. [Crossref]
15. Aleksis R, Oleskovs F, Jaudzems K, Pahnke J, & Biverstål H. Structural studies of amyloid-β peptides: unlocking the mechanism of aggregation and the associated toxicity. Biochimie, 2017, 140: 176-192. [Crossref]
16. Armstrong RA. The molecular biology of senile plaques and neurofibrillary tangles in Alzheimer’s disease. Folia Neuropathol, 2009, 47(4): 289-299.
17. Brion JP, Anderton BH, Authelet M, Dayanandan R, Leroy K, Lovestone S, et al. Neurofibrillary tangles and tau phosphorylation. Biochem Soc Symp, 2001, (67): 81-88. [Crossref]
18. Dolan PJ, & Johnson GV. The role of tau kinases in Alzheimer’s disease. Curr Opin Drug Discov Devel, 2010, 13(5): 595-603.
19. Zempel H, & Mandelkow E. Lost after translation: missorting of tau protein and consequences for Alzheimer disease. Trends Neurosci, 2014, 37(12): 721-732. [Crossref]
20. Bhat BA, Almilaibary A, Mir RA, Aljarallah BM, Mir WR, Ahmad F, et al. Natural therapeutics in aid of treating Alzheimer’s disease: a green gateway toward ending quest for treating neurological disorders. Front Neurosci, 2022, 16: 884345. [Crossref]
21. Gregory J, Vengalasetti YV, Bredesen DE, & Rao RV. Neuroprotective herbs for the management of Alzheimer’s disease. Biomolecules, 2021, 11(4): 543-553. [Crossref]
22. Kuboyama T, Tohda C, & Komatsu K. Neuritic regeneration and synaptic reconstruction induced by Withanolide A. Br J Pharmacol, 2005, 144(7): 961-971. [Crossref]
23. Ahmad M, Saleem S, Ahmad AS, Ansari MA, Yousuf S, Hoda MN, et al. Neuroprotective effects of Withania somnifera on 6-hydroxydopamine induced Parkinsonism in rats. Hum Exp Toxicol, 2005, 24(3): 137-147. [Crossref]
24. Bhattacharya A, Ramanathan M, Ghosal S, & Bhattacharya SK. Effect of Withania somnifera glycowithanolides on iron-induced hepatotoxicity in rats. Phytother Res, 2000, 14(7): 568-570. [Crossref]
25. Hussain A, Aslam B, Muhammad F, & Faisal MN. In vitro antioxidant activity and in vivo anti-inflammatory effect of Ricinus communis (L.) and Withania somnifera (L.) hydroalcoholic extracts in rats. Brazilian Archives of Biology and Technology, 2021. [Crossref]
26. Bhattacharya SK, Bhattacharya A, Sairam K, & Ghosal S. Anxiolytic-antidepressant activity of Withania somnifera glycowithanolides: an experimental study. Phytomedicine, 2000, 7(6): 463-469. [Crossref]
27. Aguiar S, & Borowski T. Neuropharmacological review of the nootropic herb Bacopa monnieri. Rejuvenation Res, 2013, 16(4): 313-326. [Crossref]
28. Valotto Neto LJ, Reverete de Araujo M, Moretti Junior RC, Mendes Machado N, Joshi RK, Dos Santos Buglio D, et al. Investigating the neuroprotective and cognitive-enhancing effects of Bacopa monnieri: a systematic review focused on inflammation, oxidative stress, mitochondrial dysfunction, and apoptosis. Antioxidants (Basel), 2024, 13(4): 393-402. [Crossref]
29. Sushma, Sahu MR, Murugan NA, & Mondal AC. Amelioration of amyloid-β induced Alzheimer’s disease by Bacopa monnieri through modulation of mitochondrial dysfunction and GSK-3β/Wnt/β-catenin signaling. Mol Nutr Food Res, 2024, 68(13): e2300245. [Crossref]
30. Shoukat S, Zia MA, Uzair M, Attia KA, Abushady AM, Fiaz S, et al. Bacopa monnieri: A promising herbal approach for neurodegenerative disease treatment supported by in silico and in vitro research. Heliyon, 2023, 9(11): e21161. [Crossref]
31. Zahara K, Bibi Y, & Tabassum S (2014). Clinical and therapeutic benefits of Centella asiatica.
32. Kandasamy A, Aruchamy K, Rangasamy P, Varadhaiyan D, Gowri C, Oh TH, et al. Phytochemical analysis and antioxidant activity of Centella asiatica extracts: an experimental and theoretical investigation of flavonoids. Plants, 2023, 12(20): 3547-3556.
33. Idris FN, & Mohd Nadzir M. Comparative studies on different extraction methods of Centella asiatica and extracts bioactive compounds effects on antimicrobial activities. Antibiotics, 2021, 10(4): 457-468. [Crossref]
34. Manmuan S, Tubtimsri S, Chaothanaphat N, Issaro N, Tantisira MH, & Manmuan P. Determination of the anticancer activity of standardized extract of Centella asiatica (ECa 233) on cell growth and metastatic behavior in oral cancer cells. Res Pharm Sci, 2024, 19(2): 121-147. [Crossref]
35. Puttarak P, Dilokthornsakul P, Saokaew S, Dhippayom T, Kongkaew C, Sruamsiri R, et al. Effects of Centella asiatica (L.) Urb. on cognitive function and mood related outcomes: a systematic review and meta-analysis. Sci Rep, 2017, 7(1): 10646. [Crossref]
36. Soumyanath A, Zhong YP, Henson E, Wadsworth T, Bishop J, Gold BG, et al. Centella asiatica extract improves behavioral deficits in a mouse model of Alzheimer’s disease: investigation of a possible mechanism of action. Int J Alzheimers Dis, 2012, 2012: 381974. [Crossref]
37. Butnariu M, Quispe C, Herrera-Bravo J, Sharifi-Rad J, Singh L, Aborehab NM, et al. The pharmacological activities of Centella asiatica L.: a review based on the mechanisms and therapeutic opportunities of its phytoconstituents. Oxid Med Cell Longev, 2022, 2022: 8214821. [Crossref]
38. Drioiche A, Ailli A, Handaq N, Remok F, Elouardi M, Elouadni H, et al. Identification of compounds of Centella asiatica by GC-MS and HPLC/UV-ESI-MS and evaluation of their antioxidant, antimicrobial, anticoagulant, and antidiabetic properties. Pharmaceuticals, 2023, 16(4): 545-556. [Crossref]
39. D’Onofrio G, Nabavi SM, Sancarlo D, Greco A, & Pieretti S. Centella asiatica L. (Saffron) in Alzheimer’s disease treatment: bioactive effects on cognitive impairment. Curr Neuropharmacol, 2021, 19(9): 1606-1616. [Crossref]
40. Siddiqui MJ, Saleh MSM, Basharuddin S, Zamri SHB, Mohd Najib MHB, Che Ibrahim MZB, et al. Saffron (Centella asiatica L.): as an antidepressant. J Pharm Bioallied Sci, 2018, 10(4): 173-180. [Crossref]
41. Meng XY, Zhang HX, Mezei M, & Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des, 2011, 7(2): 146-157. [Crossref]
42. Hospital A, Goñi JR, Orozco M, & Gelpí JL. Molecular dynamics simulations: advances and applications. Adv Appl Bioinform Chem, 2015, 8: 37-47. [Crossref]
43. Birks JS, & Harvey RJ. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev, 2018, 6(6): Cd001190. [Crossref]
44. Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, & Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res, 2009, 37(Web Server issue): W623-633. [Crossref]
45. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res, 2000, 28(1): 235-242. [Crossref]
46. White P, Hiley CR, Goodhardt MJ, Carrasco LH, Keet JP, Williams IE, et al. Neocortical cholinergic neurons in elderly people. Lancet, 1977, 1(8013): 668-671. [Crossref]
47. Francis PT, Palmer AM, Snape M, & Wilcock GK. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry, 1999, 66(2): 137-147. [Crossref]
48. Gauthier S. Advances in the pharmacotherapy of Alzheimer’s disease. Cmaj, 2002, 166(5): 616-623.
49. Eberhardt J, Santos-Martins D, Tillack AF, & Forli S. AutoDock Vina 1.2.0: new docking methods, expanded force field, and python bindings. J Chem Inf Model, 2021, 61(8): 3891-3898. [Crossref]
50. Vora J, Patel S, Athar M, Sinha S, Chhabria MT, Jha PC, et al. Pharmacophore modeling, molecular docking and molecular dynamics simulation for screening and identifying anti-dengue phytocompounds. J Biomol Struct Dyn, 2020, 38(6): 1726-1740. [Crossref]
51. Jorgensen WL, Chandrasekhar J, Madura JD, Impey R, & Klein ML. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics, 1983, 79: 926-935. [Crossref]
52. Rolta R, Salaria D, Sharma B, Awofisayo O, Fadare OA, Sharma S, et al. Methylxanthines as potential inhibitor of SARS-CoV-2: an in silico approach. Curr Pharmacol Rep, 2022, 8(2): 149-170. [Crossref]
53. Jorgensen WL, Maxwell DS, & Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. Journal of the American Chemical Society, 1996, 118: 11225-11236. [Crossref]
54. and GAK, Friesner RA, and JT-R, & Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. Journal of Physical Chemistry B, 2001, 105: 6474-6487. [Crossref]
55. Darjee SM, Modi KM, Panchal U, Patel C, & Jain VK. Highly selective and sensitive fluorescent sensor: Thiacalix[4]arene-1-naphthalene carboxylate for Zn2+ ions. Journal of Molecular Structure, 2017, 1133: 1-8. [Crossref]
56. Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res, 2000, 33(12): 889-897. [Crossref]
57. Massova I, & Kollman PA. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspectives in Drug Discovery and Design, 2000, 18: 113-135. [Crossref]
58. Patel CN, Goswami D, Jaiswal DG, Parmar RM, Solanki HA, & Pandya HA. Pinpointing the potential hits for hindering interaction of SARS-CoV-2 S-protein with ACE2 from the pool of antiviral phytochemicals utilizing molecular docking and molecular dynamics (MD) simulations. J Mol Graph Model, 2021, 105: 107874. [Crossref]
59. Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods, 2000, 44(1): 235-249. [Crossref]
60. Yang H, Lou C, Sun L, Li J, Cai Y, Wang Z, et al. ADMET SAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics, 2019, 35(6): 1067-1069. [Crossref]
61. Banerjee P, Eckert AO, Schrey AK, & Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res, 2018, 46(W1): W257-w263. [Crossref]
62. Bassani D, & Moro S. Past, present, and future perspectives on computer-aided drug design methodologies. Molecules, 2023, 28(9): 3906-3915. [Crossref]
63. LeGrand S, Scheinberg A, Tillack AF, Thavappiragasam M, Vermaas JV, Agarwal R, et al. GPU-accelerated drug discovery with docking on the summit supercomputer: porting, optimization, and application to COVID-19 research. ArXiv, 2020.
64. Chopra B, & Dhingra AK. Natural products: a lead for drug discovery and development. Phytother Res, 2021, 35(9): 4660-4702. [Crossref]
65. I. A, M. M, & I G. Neuroprotective activity of herbal medicinal products: a review. International Journal of Current Research in Physiology and Pharmacology, 2022: 1-10.
66. Ghosh AK, & Osswald HL. BACE1 (β-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem Soc Rev, 2014, 43(19): 6765-6813. [Crossref]
67. Giacobini E. Cholinesterase inhibitor therapy stabilizes symptoms of Alzheimer disease. Alzheimer disease and associated disorders, 2000, 14 Suppl 1: S3-10. [Crossref]
68. Marucci G, Buccioni M, Ben DD, Lambertucci C, Volpini R, & Amenta F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology, 2021, 190: 108352. [Crossref]
69. Grossberg GT. Cholinesterase inhibitors for the treatment of Alzheimer’s disease: getting on and staying on. Curr Ther Res Clin Exp, 2003, 64(4): 216-235. [Crossref]
70. Koly HK, Sutradhar K, & Rahman MS. Acetylcholinesterase inhibition of Alzheimer’s disease: identification of potential phytochemicals and designing more effective derivatives to manage disease condition. J Biomol Struct Dyn, 2023, 41(22): 12532-12544. [Crossref]
71. Das R, Rauf A, Akhter S, Islam MN, Emran TB, Mitra S, et al. Role of Withaferin A and its derivatives in the management of Alzheimer’s disease: recent trends and future perspectives. Molecules, 2021, 26(12): 3696-3703. [Crossref]
72. Khan SA, Khan SB, Shah Z, & Asiri AM. Withanolides: biologically active constituents in the treatment of Alzheimer’s disease. Med Chem, 2016, 12(3): 238-256. [Crossref]
73. Szutowicz A, Bielarczyk H, Jankowska-Kulawy A, Ronowska A, & Pawełczyk T. Retinoic acid as a therapeutic option in Alzheimer’s disease: a focus on cholinergic restoration. Expert Rev Neurother, 2015, 15(3): 239-249. [Crossref]
74. van de Waterbeemd H, & Gifford E. ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov, 2003, 2(3): 192-204. [Crossref]