Open Access | Review
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
Diverse models in Alzheimer's research: exploring alternative approaches beyond traditional rodent frameworks
# These authors contributed equally.
* Corresponding author: Govind Singh
Mailing address: Department of Pharmaceutical Sciences, Maharshi Dayanand University, Rohtak, 124001, India.
Email: drgovind.pharma@mdurohtak.ac.in
Received: 19 November 2024 / Revised: 04 December 2024/ Accepted: 13 December 2024 / Published: 28 December 2024
DOI: 10.31491/APT.2024.12.160
Abstract
Alzheimer's disease (AD) is a debilitating neurodegenerative disorder that progressively impairs cognitive function and memory. It is characterized by the buildup of beta-amyloid plaques and neurofibrillary tangles, leading to the progressive destruction of brain cells, especially in the hippocampus and neocortex. Despite extensive research, the precise molecular mechanisms underlying AD remain unclear. Rodent models, that are the mainstay of AD research, offer inherent limitations, thus prompt the search and exploration of alternative models. To address this knowledge gap, scientists have turned to non-mammalian models such as zebrafish, fruit flies, and worms. These organisms provide a valuable platform for studying AD due to their shorter lifespans, ease of genetic manipulation, and lower maintenance costs. Additionally, they facilitate high-throughput screening and real-time imaging, which accelerates the investigation of underlying pathogenesis and discovery of potential drug targets. By exploring the complex pathogenesis of AD using these models, researchers aim to develop innovative therapies to combat this devastating disease.
Keywords
Alzheimer's disease, Drosophila melanogaster (fruit flies), Caenorhabditis elegans (worms), Danio rerio (zebra fish), transgenic models, neurodegeneration
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder (NDD) characterized by a gradual decline in memory, learning, and
cognitive abilities. It is the leading cause of dementia, a condition marked by the
deterioration of cognitive function [1]. AD poses a significant and growing public health concern. It currently affects
over 6 million Americans aged 65 and older, with projections
indicating that this number could more than double by 2060 in the absence of significant medical breakthroughs
[2]. Tragically, AD has become a leading cause of death worldwide, with
mortality rates escalating dramatically over the past two decades, a trend likely exacerbated by the COVID-19
pandemic [3]. According to data from the GBD study 2021, there was an
elevation in AD prevalence by 160.8% from 1990 to 2021 [4].
Despite advancements in symptom management, a definitive cure for AD remains elusive. Discovering effective
treatments for AD and other NDDs is a paramount challenge of the 21st century [5,
6] AD is indeed defined by the abnormal accumulation of proteins
in the brain, which ultimately leads to neuronal death and tissue degeneration
[7]. The underlying causes of AD are complex and multifaceted, involving
impaired communication between brain cells, oxidative stress, mitochondrial dysfunction [8],
apoptosis [9], inflammation
[10], and aberrant brain signaling [11].
Current treatments can only alleviate symptoms and do not halt disease progression
[7]. Recent advancements in human genetics have
significantly enhanced our comprehension of the genetic underpinnings of NDDs, including Parkinson's
disease and AD. However, ethical considerations and technical limitations impede human-based research. Consequently, animal
models, primarily rodents, have become indispensable tools for AD research. While these models
can recapitulate certain AD characteristics, such as neuronal alterations and behavioral abnormalities, they fall short of fully
capturing the disease's complexity [12-14].
Rodent models, despite their contributions, exhibit substantial limitations, including inadequate
replication of human AD, extended lifespans, slow disease progression, ethical concerns, and high
resource demands. To circumvent these challenges, researchers have turned to non-mammalian models like zebrafish, fruit flies, and
nematodes [15-17].
Characterized by shorter lifespans and lower maintenance requirements, these organisms offer advantages for investigating the
fundamental mechanisms of AD. Moreover, the development of numerous transgenic strains
expressing both human and complementary amyloid-β (Aβ) sequences in non-rodent models has expanded research possibilities, yielding strains
that produce either Aβ1-42 or Aβ3-42
[18-21].
Table 1 highlights the key advantages of these novel models over the traditional rodent model for AD research.
Table 1
Comparison between rodent and non-rodent models for AD research.
| Features | Rodent model | Non-rodent model (C.elegans, Drosophila, and zebrafish) |
|---|---|---|
| Complexity of nervous system | Highly complex, with a brain and spinal cord | Relatively simple nervous system |
| Genetic manipulation | Advanced genetic techniques are required for genetic manipulatio | Highly amenable to genetic manipulation |
| Lifespan | Months to years | Days to week |
| Cost | Relatively high cost, due to housing, feeding, and experimental procedures | Low cost, due to simple housing and feeding requirements |
| Ethical considerations | Significant ethical considerations | Minimal or no ethical considerations |
| Behavior and cognition | Complex | Simples |
The nematode, Caenorhabditis elegans (C. elegans), has emerged as a powerful model organism for dissecting the underlying mechanisms of human NDDs, including AD [19, 20, 22]. Its rapid lifespan, microscopic size, and economical maintenance render it highly efficient for research [23]. Notably, C. elegans possesses numerous human gene orthologs, including those implicated in AD, as shown in Figure 1. This genetic homology, coupled with well-established neurodegenerative pathway analysis techniques, positions the nematode as an ideal system for exploring the cellular and molecular intricacies of AD [24, 25].
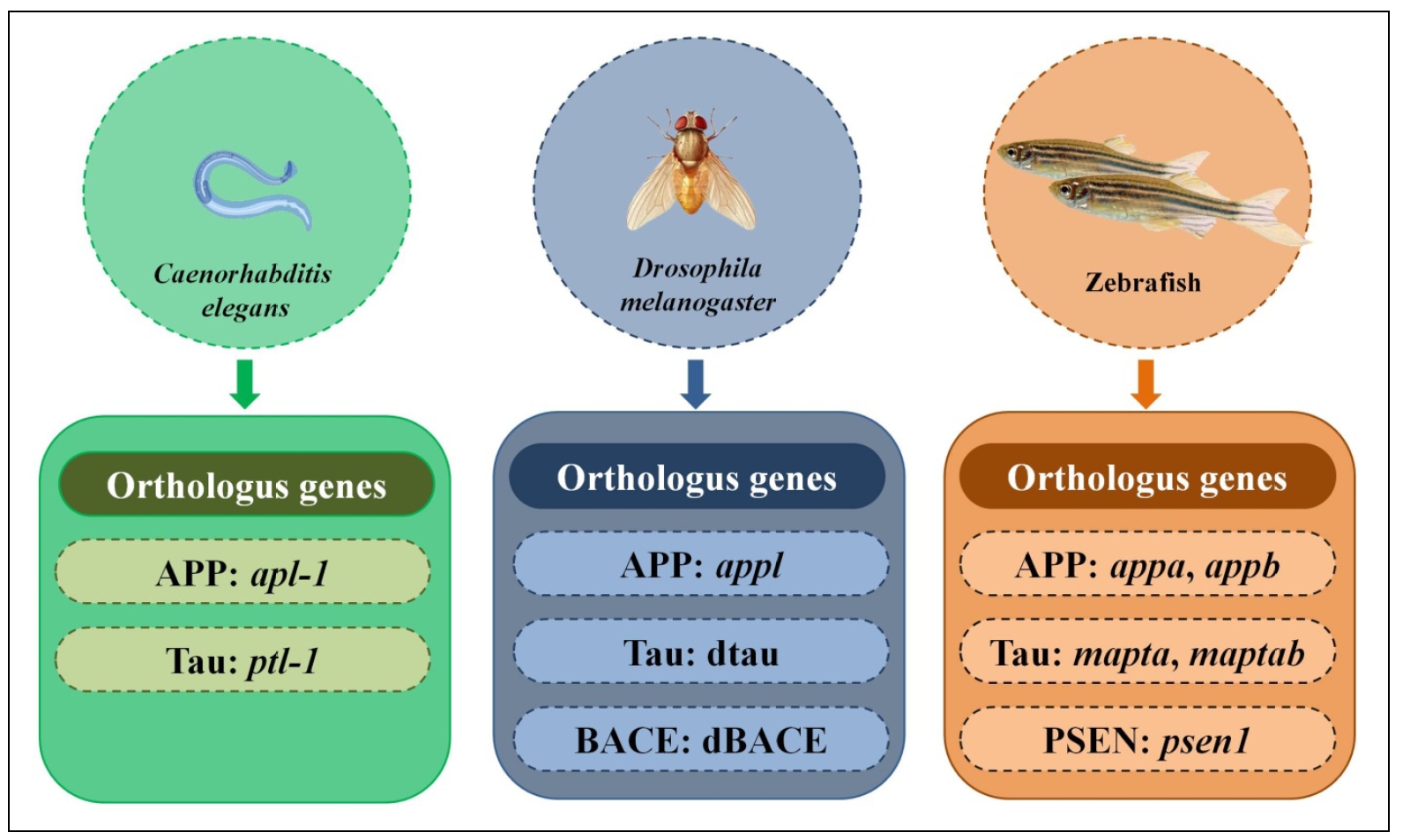
Figure 1. Non-rodent models and their orthologous genes with humans.
For over a century, Drosophila melanogaster (fruit fly), has been a fundamental tool in biological research,
significantly advancing our understanding of genes, chromosomes, and genetic inheritance [26].
Beyond its genetic contributions, Drosophila has proven to be a valuable model organism for investigating
human NDDs at molecular, physiological, and behavioral levels [27,
28]. It's simple yet conserved anatomy and genetic framework make it an
ideal system for unraveling complex disease mechanisms [29].
Drosophila research has led to groundbreaking discoveries regarding critical AD-related processes and
pathways, and numerous models have been established to study
amyloidosis and tauopathy [26].
The zebrafish has also emerged as a valuable vertebrate model organism for investigating human NDDs, particularly
AD [30, 31]. Its relatively
simple yet conserved nervous system facilitates detailed imaging and analysis of neuronal proteins, providing
unparalleled insights into neurological processes. Notably, zebrafish exhibit behavioral patterns strikingly similar
to humans, suggestingshared neural mechanisms underlying behavior such as learning, social interactions, and
decision-making [28]. Researchers have successfully developed zebrafish
models that recapitulate key pathological features of AD, including tauopathy and amyloid-beta accumulation.
These models offer a powerful platform for elucidating disease mechanisms and identifying potential therapeutic
targets. As a result, the zebrafish is rapidly gaining prominence as a cornerstone in neuropharmacological
research [32-34].
This article highlights the increasing significance of nonrodent animal models, particularly zebrafish,
fruit fly, and C. elegans, in AD research. These models offer distinct advantages over traditional rodent models,
such as accelerated development timelines, reduced costs, and enhanced capacity for high-throughput screening,
as illustrated in Figure 2. Zebrafish, with their transparent embryos and
comprehensively characterized nervous system, are uniquely suited for studying neuronal development and
function. C. elegans, a genetically tractable nematode, provides a robust toolkit for dissecting complex biological
processes. By harnessing the strengths of these models, researchers can expedite the discovery and development
of novel therapeutic interventions for AD and other neurodegenerative diseases. These invertebrate systems serve as
invaluable platforms for elucidating disease mechanisms and identifying promising drug targets.
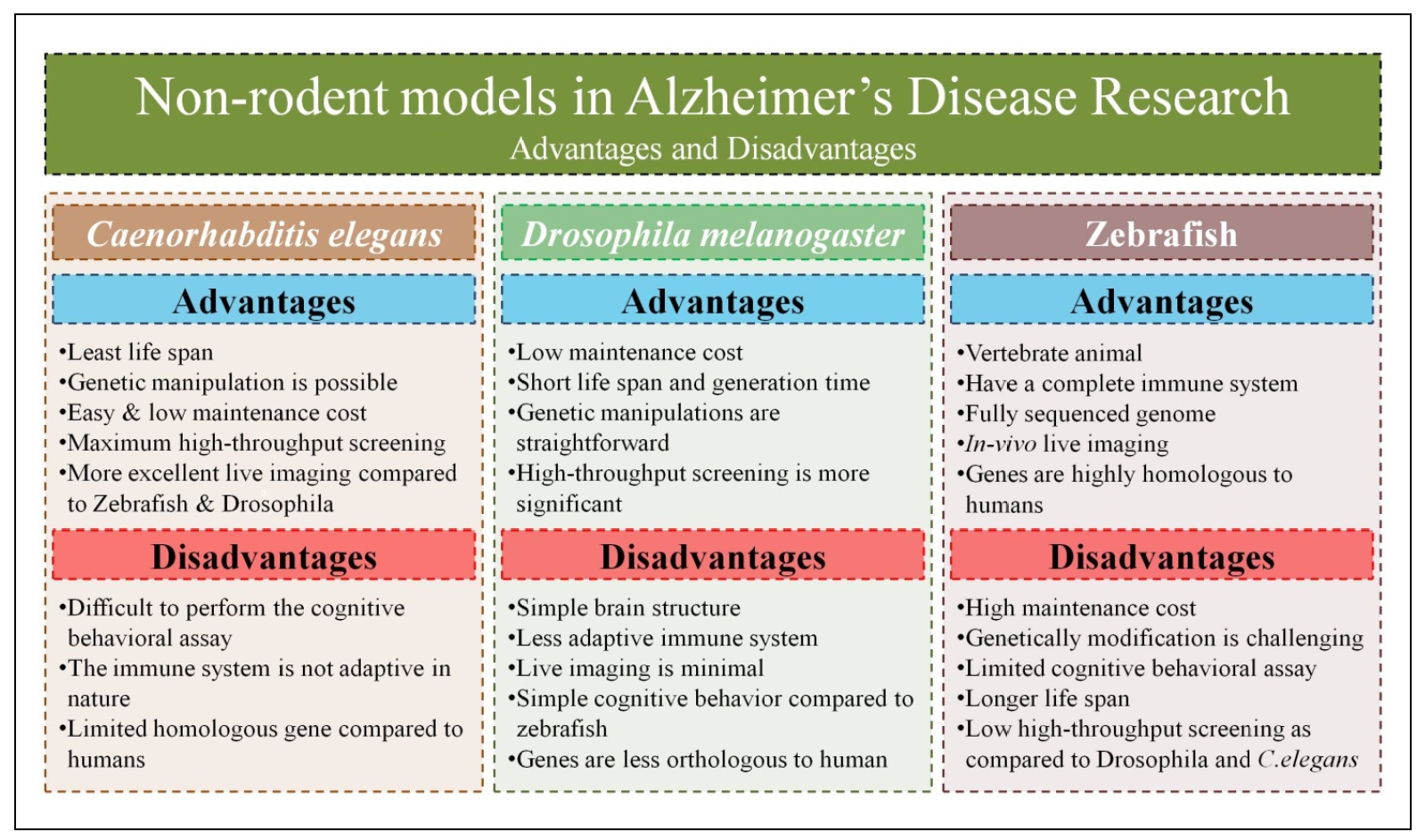
Figure 2. Advantages and disadvantages of various alternatives to rodent models in AD research.
Caenorhabditis elegans as a model for AD
C. elegans, a microscopic nematode, has emerged as a valuable model organism for studying AD
and other NDDs. They were introduced by Sydney Brenner in 1963, its simple anatomy, transparent body, and short lifespan make
them highly amenable to genetic and biochemical studies [35]. The complete
sequencing of the C. elegans genome revealed striking similarities to the human genome, including
shared genes involved in AD pathogenesis, such as APP and tau. This organism offers several advantages over
other in vivo models. Its relatively small nervous system enables detailed investigation of neuronal processes,
while its transparent body facilitates realtime observation of cellular events [36,
37]. Additionally, C. elegans is highly amenable to
genetic manipulation, allowing for the creation of transgenic lines and highthroughput screening, which have
been instrumental in elucidating the functions of AD-related genes and proteins [38,
39].
C. elegans provides a valuable platform for understanding the molecular mechanisms underlying AD.
While lacking the Aβ sequence, the nematode possesses a single amyloid precursor protein (APP) homolog, apl-1,
which shares structural similarities with the human APP family [40].
Although lacking a clear BACE ortholog, C. elegans harbors ADAM10 and ADAM17 homologs,
enzymes involved in APP processing [41, 42].
A transmembrane motif as well as a small cytosolic region with 71% sequence identity to the
mammalian APP is also present. Notably, apl-1 lacks the Aβ sequence in contrast to APP but is otherwise
comparable to APLP1 and APLP2 [43,
44]. The sup-17 and adm-4 genes throughout C. elegans
are present and resemble the human ADAM10 and ADAM17 genes, respectively [45].
Bioinformatic investigations have not shown the existence of a BACE ortholog or other β-secretase activities
that break down mammalian APP into C. elegans [46].
Human PSEN1 and PSEN2 also known as S182 and E5-1 respectively, were initially characterized
as new proteins with numerous transmembrane domains [47].
Presenilins, key components of the γ-secretase complex implicated in AD, are conserved between humans
and C. elegans [46]. The two
C. elegans Notch genes, lin-12 and glp-1, are implicated in germline development and
various cell fate decisions throughout development and are also included in vulvae cell specifications
[48]. The nematode sel-12 protein is the functional homolog
of human presenilins and is essential for Notch signaling, a pathway crucial for development. The ability to rescue
loss-of-function sel-12 mutations with human presenilins underscores the functional conservation
of these proteins [49, 50].
Additionally, C. elegans possesses additional components of the γ-secretase complex, including
Aph-1, Aph-2/NCT, and pen-2, which interact with sel-12/psen to facilitate Notch
signaling [51]. Expanding on the role of presenilin homologs in
C. elegans, spe-4, sel-12, and hop-1 were also identified as members of the PSEN gene
family. Notably, sel-12 exhibits a higher degree of sequence homology to mammalian PSEN, suggesting a
conserved role in regulating APP processing. Overexpression of apl-1 in C. elegans led to
increased mortality, which could be rescued by sel-12 variants, implicating sel-12 in the
control of apl-1 cleavage. Furthermore, sel-12 mutations in the nematode have been associated with
disruptions in mitochondrial calcium homeostasis, a hallmark of AD pathology
[52, 53]. These genetic and biochemical similarities between humans
and C. elegans make the nematode a powerful model for investigating the molecular basis of AD and for identifying potential therapeutic targets.
Aβ and tau in C. elegans
The progressive nature of AD is reflected in animal models such as earthworms expressing mammalian
Aβ3-42 exhibited age-dependent paralysis, with a more rapid and severe onset observed with Aβ1-42
expression [54, 55].
Similarly, C. elegans exposed to aggregated Aβ display muscle paralysis and neuronal toxicity.
This model system offers a platform for identifying molecules and pathways capable of inhibiting Aβ
aggregation or promoting disaggregation [56]. Researchers have demonstrated that
C. elegans extracts possess the ability to disassemble aggregated human Aβ3-42,
independent of protease activity, suggesting the involvement of novel protein factors in this process
[57]. The C. elegans model has proven invaluable in
identifying potential therapeutic agents capable of disaggregating Aβ. High-throughput drug screening using this model has
accelerated the discovery process [58]. For instance, PBT2,
an analog of 8-hydroxyquinoline, demonstrated remarkable efficacy in reversing AD-like symptoms in mice within days. This
compound also prevented paralysis in C. elegans expressing inducible Aβ1-42,
highlighting its potential to target Aβ-induced neurotoxicity [59].
C. elegans offers insights into the role of heat shock chaperone proteins in modulating Aβ
toxicity. Researchers have identified several C. elegans orthologs of human heat shock chaperone proteins that interact
directly with Aβ3-42 [60]. Notably, B-crystallin and
HSP-16 proteins, including HSP-16-48, HSP-16-1, and HSP-16-2, bind to both intracellular Aβ3-42 monomers and
soluble oligomers, but not fibrillar forms [61]. Interestingly,
Aβ3-42 transgenic lines exhibit increased hsp-16-2 transcript levels, suggesting a potential role for
this chaperone in response to Aβ stress. However, the precise function of these chaperones in promoting
or inhibiting Aβ-induced paralysis remains to be elucidated [62]. In contrast to
the HSP-16 family, another study by Fonte et al. demonstrated a protective role for the HSP70
chaperone in preventing paralysis induced by Aβ in C. elegans. These findings align with human studies reporting
increased levels of both HSP70 and B-crystallin in AD brains, suggesting a potential role for these
chaperones in binding and mitigating Aβ toxicity [63-65].
The clearance of Aβ from the brain is a also critical process linked to apolipoprotein E (ApoE) and the
LRP2/megalin receptor [66]. Yochem
and their team have discovered LRP-1 in the nematode, a protein that shares significant structural
and functional similarities with the human protein LRP2/megalin. This homology makes C. elegans an ideal
organism for exploring the role of LRP-2/megalin-like functions in these pathways [67].
Neurofibrillary tangles (NFTs), primarily composed of hyperphosphorylated tau protein, are also a hallmark of AD.
C. elegans possesses a single tau ortholog, ptl-1, which shares structural similarities with
human tau, including multiple tau protein-like repeats. The two main isoforms, ptl-1a and ptl-1b,
exhibit high sequence homology to the C-terminal region of human tau. Like human tau, C. elegans ptl-1 binds
to microtubules and promotes tubulin polymerization in vitro [68].
The C. elegans protein ptl-1, similar to human tau, shows changing distribution patterns throughout
its development. Initially present in the head nerve cells and
growing outer layer of embryos, ptl-1, is later detected in most neurons that respond to light touch
[69]. Notably, loss-of-function mutations in ptl-1 result in
embryonic lethality, mirroring the phenotype observed with ptl-1 overexpression, suggesting a critical
role for this protein in early developmental processes [68]. While
ptl-1/tau mutant animals that survive embryogenesis exhibit seemingly normal microtubule organization
at the light microscopic level, they display significant deficits in gentle touch sensitivity compared to wild-type
controls. Despite normal growth and development, these mutants exhibit reduced lifespan, suggesting a broader
impact of ptl-1 dysfunction on organismal health and longevity [70].
Transgenic C.elegans models for AD
In an effort to unravel the complexities of AD, researchers have developed a diverse range of C. elegans
strains that carry specific genetic alterations. These engineered worms serve as valuable models to study the
underlying mechanisms of NDDs [60]. Treusch and colleagues identified
several neurodegenerative modifiers related to the cytoskeleton, including
GRR1, YAP 1802, SLA1, KEM1, CRM1, INP52, and RTS,
which offer potential targets for therapeutic intervention [71,
72]. A
significant milestone in AD research occurred in 1995 with the creation of the CL2006 strain, the first
C. elegans transgenic model to express human Aβ1-42 in body wall muscle. This model has become a crucial
platform for testing the therapeutic potential of various herbal and synthetic compounds
[71, 72]. Furthermore, Diomede and colleagues tested
the effects of silver nanoparticles on transgenic C. elegans producing amyloidogenic proteins.
These worms, specifically strains CPV10 and CL2120, mimic diseases caused by the buildup of Aβ, a substance linked to many
age-related illnesses. The researchers found that silver nanoparticles harm both normal and transgenic
C. elegans in various ways. To gauge the sensitivity of the worms, they compared their results to those obtained
using common mammalian cell lines, D384 brain cells and A549 lung cells [73].
Additionally, he and colleagues evaluated seven newly developed compounds (2-aryl ethenyl quinolines) for
their ability to protect nerve cells using the CL2355 strain of C. elegans as an AD model. Two
of these compounds improved memory problems and reduced the buildup of harmful Aβ clumps in these worms
[74]. Nevertheless, a variety of C. elegans transgenic
models have been generated to study AD, including UA166, CL2355, CL2122, CL2006, and CL4176
[75, 76]. Despite significant
advancements in transgenic C. elegans models of AD, a critical gap remains. No existing strain displays
constitutive, pan-neuronal expression of Aβ1-42, nor does it accurately replicate the age-dependent
behavioral deterioration that characterizes human AD. This limitation hinders the ability to fully understand the
complex mechanisms underlying the disease and develop effective therapeutic strategies. Recently, by utilizing
transgenic AD model strain (GRU102), which expresses pathogenic human Aβ1-42 exclusively in neurons, Toe
and team successfully identified metformin, lithium, and curcumin as promising candidates for AD treatment
[77].
In AD, tau proteins undergo changes in structure and function. To study these changes and their effects, Brandt
and colleagues created C. elegans worms with human tau proteins. These worms were made in two versions: one
with normal tau and another with a modified, abnormally structured tau (pseudo-hyperphosphorylated tau). Both types
of worms showed similar movement problems. However, only the worms with the abnormal tau developed clumps of tau
protein and had problems with motor neuron development [78]. Three
different C. elegans models—myo-3, snb-1, and unc-54—are commonly used to test how well drugs protect nerve
cells from damage caused by Aβ. The unc-54 model, where Aβ builds up inside cells, offers a unique opportunity to
identify cellular proteins that interact with Aβ. These proteins might be involved in either causing Aβ-related
damage or protecting the cell from it [63].
C.elegans AD models for screening of new therapeutic agents
Several natural and synthetic compounds have shown promise in combating AD when tested on C. elegans models [79, 80]. McCormick and colleagues tested azaperone, an antipsychotic drug that blocks dopamine D2 receptors, in a C. elegans model of AD. They found that this drug improved the worms' movement and decreased the levels of abnormal tau protein. These results suggest that blocking dopamine D2 receptors might be a promising approach to preventing nerve damage caused by tau protein [81]. Additionally, caffeic acid has shown promising results in combating the harmful effects of Aβ in CL4176 C. elegans worms. This compound significantly reduced Aβ toxicity, extended lifespan, improved reproductive issues, and alleviated body paralysis. It also activated protective heat-shock proteins by increasing hsf-1 and hsp-16.2 gene activity [82]. In another study, researchers examined the effects of magnolol on Aβ-related diseases using transgenic C. elegans worms producing human Aβ. They discovered that magnolol can activate PPAR-gamma, boost ApoE levels, and enhance the ability of microglia cells to clear Aβ, thereby reducing Aβ-induced toxicity and memory impairment [83]. Further the water extract from Terminalia chebula reduced Aβ clumps and improved paralysis in AD model worms (CL2006 and CL4176) [84]. Nicotine also lessened paralysis symptoms in CL2120 and CL4176 models by decreasing Aβ buildup and harmful oligomers, although it did not prevent Aβ clumping [85]. Ginkgo biloba extract (EGb 761) and its component ginkgolide A improved various AD-related behaviors in C. elegans by reducing Aβ clumping and harmful oligomers [86]. Finally, oleuropein, when tested in CL2006 worms, reduced Aβ plaque buildup, Aβ oligomers, and paralysis while extending lifespan [87]. Recently, it was observed that ethyl caffeate treatment significantly delayed the onset of paralysis in the CL4176 C. elegans model of AD, following the reduction in the severity of 5-hydroxytryptophan (5-HT) induced paralysis, suggesting a neuroprotective effect. Furthermore, it also activated the nuclear translocation of the transcription factors DAF-16 and SKN-1, leading to increased expression of the stress response genes sod-3, gst-4, and hsp-16.2 [88]. Hence, these findings suggest that natural compounds may offer potential therapeutic targets for AD.
Drosophila melanogaster as a model for AD
Drosophila melanogaster has emerged as a cornerstone model organism in biological research, particularly for understanding complex human diseases [27, 28]. With a rich genetic history spanning over a decade, this fruit fly offers a unique combination of scientific, genetic, behavioral, anatomical, and economic advantages. Its genome shares remarkable homology with humans, with nearly two hundred Drosophila genes corresponding to human disease-associated genes [29]. The simpler genetic makeup of Drosophila compared to vertebrates facilitates gene characterization, while its less complex but structurally similar brain enables the study of NDDs. Furthermore, Drosophila exhibits age-related behavioral changes and physiological mechanisms, such as oxidative stress, that mirror aspects of human NDDs [89, 90]. These combined attributes make Drosophila an invaluable tool for investigating the underlying biology of complex disorders and identifying potential therapeutic targets [26]. It has proven to be a valuable model for studying AD due to its genetic simplicity, rapid life cycle, and conserved biological pathways. With a fully sequenced genome of about 30,600 protein-coding genes spread across four chromosomes, Drosophila offers exceptional genetic tractability. Its well-characterized nervous system, including the complex eye, allows for detailed phenotypic analysis. The fly exhibits a range of behaviors, from simple reflexes to complex learning and memory, which can be easily quantified [91-93]. This model of AD has been created by overexpressing human AD-related genes like tau and Aβ42, resulting in neuronal decline and mimicking key pathological features of AD. Observable phenotypic changes, such as impaired flight, reduced motility, blindness, and early death, provide clear endpoints for assessing disease progression. Additionally, Drosophila models have shown memory deficits, a hallmark of AD. This ability to study both cellular and behavioral aspects of neurodegeneration has greatly advanced our understanding of AD pathogenesis [94, 95].
Aβ and tau in Drosophila model
Comparative genomics reveals striking similarities between human and Drosophila gene structures, providing a
strong foundation for using Drosophila as an AD model [96].
A significant proportion of human genes associated with AD have homologous counterparts in Drosophila.
While Drosophila possesses a β-secretase-like enzyme, its activity is notably low
[97-99]. However, the fly's
APP homolog, dAPPl, shares structural similarities with its vertebrate counterpart. Overexpressing the Drosophila
β-secretase-like enzyme can lead to the cleavage of dAPPl and the generation of peptides resembling human Aβ
peptides. Intriguingly, the accumulation of these peptides in
Drosophila is linked to neurodegeneration and age-related behavioral deficits. These findings underscore
the potential of Drosophila as a valuable tool for understanding the molecular mechanisms underlying AD
pathogenesis [100]. In addition to Aβ42 plaques,
hyperphosphorylated tau protein, and the resulting NFTs are key pathological features of AD. While the interaction between
these two hallmark features is not fully understood, Drosophila models expressing Aβ42
have been used to explore whether endogenous tau can form fibrillary aggregates [101].
Despite the presence of Aβ42, these models did not show hyperphosphorylated tau or NFT formation.
On the other hand, overexpressing both wild-type and mutant human tau
in Drosophila resulted in intracellular lesions and hyperphosphorylation, though classic filamentous
structures were not observed. Nevertheless, these tau-expressing flies exhibited neurodegeneration and abnormal tau
protein distribution, as shown by immunostaining [27,
102]. Additionally, the development of vacuolar lesions in the
Drosophila nervous system, along with age-dependent neurodegeneration and early mortality, highlights
the model's value for studying tau-related pathologies [103]. To
investigate the synergistic effects of tau and Aβ42, Folwell and colleagues co-expressed both
proteins in Drosophila. The combination of these pathological proteins exacerbated tau-induced axonal transport
defects, reduced survival, and neuronal dysfunction. This model system provides a valuable platform
for studying the early stages of AD pathogenesis and identifying potential genetic modifiers
[104]. Utilizing the rough eye phenotype as a screening tool,
researchers identified Drosophila homologs of GSK-3β as modulators of tau-mediated toxicity. Overexpression of
GSK-3β intensified tau-induced eye defects, while its reduction had a protective effect. Mechanistic
studies revealed that GSK-3β plays a critical role in promoting pathogenic tau phosphorylation [105].
To elucidate the relationship between tau phosphorylation and toxicity, researchers have generated
Drosophila models expressing tau variants resistant to specific phosphorylation sites. By mutating serine or threonine
residues to alanine, Chatterjee et al. created fly lines expressing tau proteins resistant to phosphorylation
by kinases such as GSK-3β and PAR-1. This approach allowed for a detailed examination of the role of specific
phosphorylation sites in tau-mediated toxicity. Interestingly, while the overexpression of GSK-3β exacerbated
the rough eye phenotype in flies expressing wild-type tau, it had a less pronounced effect on flies expressing the
TauS2A mutant, despite increased overall tau phosphorylation. Conversely, the TauS11A mutant, which was resistant
to GSK-3β phosphorylation, exhibited an exacerbated rough eye phenotype when GSK-3β was overexpressed
[106]. Furthermore, building on previous research,
Iijima-Ando et al. generated an additional Tau variant, TauS262A, which is resistant to phosphorylation
at serine 26 [107]. Similarly, Steinhilb and team unveiled the critical
role of phosphorylation in tau-mediated neurotoxicity. The creation of phospho-deficient (TauAP) and phospho-mimetic (TauE14)
Tau variants provided compelling evidence that altered phosphorylation states can significantly
impact disease progression [108]. These findings suggest a complex interplay between tau
phosphorylation, specific phosphorylation sites, and toxicity. Moreover, the study revealed no consistent
correlation between tau's microtubule binding properties, aggregation, and the rough eye phenotype, indicating that other
factors may contribute to tau-mediated toxicity.
Role of metal ion in AD by using the Drosophila model
Drosophila models have also been pivotal in exploring the impact of metal ions on AD pathogenesis [109]. Excessive aluminium, cadmium, lead and zinc exposure leads to neurodegenerative symptoms such as shortened lifespan, impaired locomotion, olfactory learning deficits, and brain degeneration [110-114]. The complex relationship between iron and Aβ peptides has been a key area of investigation. Iron has been found to influence Aβ42 toxicity, with iron chelation mitigating Aβ42-induced damage. Interestingly, while ferritin knockdown protected against Aβ42 toxicity, it did not impact Aβ accumulation. Biophysical studies have shown that Fe2+ affects Aβ fibril formation,indicating a complex interaction between iron and Aβ in disease progression [115]. Research has demonstrated that dietary supplementation with copper or zinc exacerbates Aβ42-induced neurotoxicity in flies, resulting in a reduced lifespan and locomotors abnormalities [116-119].
Transgenic Drosophila model for AD
To investigate endogenous Aβ production and human Aβ42-induced neurotoxicity, several transgenic Drosophila models have been developed, expressing variant genes associated with familial AD [120]. Greeve et al. created a triple transgenic fly expressing human APP and human β-secretase, incorporating familial AD mutations (N141I, L235P, and E280A). These flies exhibited age-dependent neurodegenerative phenotypes including photoreceptor cell loss, severe axonal degeneration, and premature death [121]. The co-expression of human APP and β-secretase in Drosophila facilitated the production of a highly glycosylated form of human APP, leading to the formation of Aβ40 and Aβ42 plaques in transgenic tissues. This triple transgenic model effectively recapitulates key metabolic processes associated with Aβ42 accumulation in humans, highlighting the utility of Drosophila for studying AD pathogenesis [122]. To circumvent the complexities of APP processing, researchers have developed Drosophila models expressing fully processed Aβ peptides. Finelli et al. generated transgenic fly lines expressing human Aβ40 and Aβ42, enabling the investigation of Aβ accumulation and toxicity in various cell types, including brain cells. Notably, Aβ42, but not Aβ40, accumulated in the Drosophila brain, leading to dose- and age-dependent neurodegeneration, cognitive deficits, locomotor impairments, and reduced lifespan [123]. Furthermore, Crowther et al. employed a more direct approach by fusing Aβ40/42 peptides to a native Drosophila secretory signal sequence. Using the UAS/Gal4 system, these researchers achieved spatiotemporal control over Aβ40/42 expression, bypassing the influence of APP processing. This model offers a streamlined approach to studying the toxic effects of Aβ peptides independently of APP-related factors [120]. Although Drosophila does not possess a native APOE gene, Haddadi et al. developed a groundbreaking transgenic model by introducing human APOE. This innovative approach allows for the investigation of neurodegenerative disease mechanisms and potential therapeutic interventions in a genetically tractable organism [124].
Drosophila AD model for screening of new therapeutic agents
Recent studies have unveiled a complex interplay between epidermal growth factor receptor (EGFR) and Aβ42 in Drosophila models of AD. Overexpression of EGFR exacerbated Aβ42-induced cognitive decline, suggesting a detrimental role for EGFR in AD pathology. Conversely, EGFR inhibitors, gefitinib and erlotinib, commonly used in cancer therapy, effectively ameliorated Aβ42-associated memory loss in Drosophila. These findings highlight the potential therapeutic benefits of targeting EGFR in AD. Additionally, the study demonstrated the efficacy of memantine, a standard-of-care AD drug, in mitigating Aβ42-induced cognitive impairments in flies, further validating the Drosophila model for AD research [125]. Several natural compounds have shown promise in combating AD such as Curcumin, a compound derived from turmeric, has shown promise as a potential therapeutic for AD in Drosophila models. When administered to flies expressing Aβ42, curcumin significantly improved lifespan by up to 75% and enhanced locomotor activity. Mechanistic studies suggest that curcumin promotes the conversion of Aβ oligomers (toxic species) into less harmful amyloid fibrils, thereby reducing neurotoxicity [126]. The therapeutic potential of a novel compound, XJP-1, has been explored using the Aβ-arc transgenic Drosophila model of AD. Derived from a substance found in Musa sapientum L. peel, XJP-1 demonstrated promising effects in ameliorating AD-related symptoms in flies. Treatment with XJP-1 led to improved locomotor function, reduced amyloid plaque formation, and extended lifespan [127]. Further, acacetin, isolated from Agastache rugosa has shown promise by suppressing APP synthesis and Aβ production, potentially through downregulation of APP and BACE-1 mRNA [128]. D737, another compound, extended lifespan and improved locomotor activity in AD flies by inhibiting Aβ42 aggregation and reducing oxidative stress [129]. Additionally, Quercetin, a flavonoid, has also demonstrated therapeutic potential in Drosophila AD models by mitigating Aβ toxicity. It regulates the expression of cell cycle-related genes, restoring cyclin B levels and ultimately improving lifespan and climbing ability [130]. These findings collectively highlight the potential of natural compounds as promising candidates for AD drug development.
Zebrafish as a model for AD
In the 1970s, George Streisinger at the University of Oregon pioneered the use of zebrafish as a model organism,
noting their simpler biology and easier genetic manipulation compared to mice. Over time, zebrafish models have
proven crucial in dissecting the complex mechanisms connecting AD features, such as tau and Aβ buildup,
to neuronal damage and cognitive decline [131,
132]. While rodent models have also been valuable in AD research,
zebrafish have provided unique insights into genes linked to AD that were challenging to study before. Despite
significant progress in understanding AD through zebrafish research, the exact molecular processes driving
neurodegeneration are still not fully understood [34,
133]. Nonetheless, zebrafish have become a fundamental tool in
biomedical research in the past decade, with applications extending beyond NDDs
[134]. They allow for precise genetic manipulation at physiologically
relevant levels, exceeding the capabilities of rodents. Their relatively large size, accessibility, and transparent
embryos make it easy to observe gene function during development. As a result, zebrafish embryos are an
invaluable vertebrate model for investigating the cellular and molecular mechanisms associated with AD-related
genes [135, 136]. Although
many zebrafish genes are recognized as orthologs of human genes, the complete scope of genetic homology is still being
explored.
Zebrafish exhibit a brain structure strikingly similar to that of mammals, including key neurotransmitter
systems and a functionally comparable blood-brain barrier. Many genes associated with AD in humans have direct
counterparts in zebrafish, such as the "co-orthologs" appa and appb, which are analogous
to the human APP [137-139]. The
zebrafish model is particularly suited for genetic manipulation. Techniques such as mRNA injection, genome
editing, and transgenesis provide precise control over gene expression, allowing researchers to study both subtle and
significant changes in gene function during development [140].
Combined with the transparency of zebrafish embryos, these features offer exceptional opportunities for observing the
cellular and molecular impacts of genetic alterations.
Zebrafish exhibit striking similarities to humans in terms of brain structure and function, making
them a valuable model for studying neurological diseases. Key proteins involved in AD, such as β-secretase and tau, have
identifiable counterparts in zebrafish [141-143].
β-secretase and tau, both species share similar brain regions, neurotransmitter systems, and
cellular components [144-146].
Additionally, they have enzymes for production and metabolism, as well as neurotransmitters such as glutamate,
serotonin, histamine, GABA, dopamine, and acetylcholine [147,
148]. Astrocytes, microglia, oligodendrocytes, motor neurons, cerebellar
Purkinje cells, and myelin present in zebrafish have characteristics that are also similar to those of human cells,
demonstrating that similarities exist even at the cellular level
[149-151]. Furthermore, zebrafish
display comparable behavioral patterns and learning capabilities to humans. Researchers have demonstrated
that zebrafish exposed to Aβ1-42, a hallmark peptide of AD, develop cognitive impairments and
increased tau phosphorylation, mimicking key features of the human disease [133].
Researchers also have looked into non-associative learning throughout zebrafish larvae according to the
cognitive and behavioral reactions of AD. Larvae have been subjected to a
wide range of different stimuli seven days post fertilization (dpf) and they significantly reduced
their startle response [152]. Indeed, another group administered Aβ1-42
through injection to zebrafish embryos that were 24 hours post-fertilization (hpf). They noticed significant
cognitive impairments in the 5 dpf larvae of the embryos having higher tau hyper-phosphorylation in
GSK3 targeted residues [153]. These findings underscore the
potential of zebrafish as a model organism for understanding the underlying mechanisms of AD and exploring therapeutic
interventions.
Zebrafish have also emerged as valuable models for studying the vascular and neurodegenerative aspects
of AD [154]. Research has shown that elevated levels of Aβ in zebrafish can induce
abnormal blood vessel growth, while Aβ deficiencies lead to cerebral hemorrhage. Interestingly,
the administration of human Aβ1-42 can reverse these hemorrhages, suggesting a complex role for Aβ in cerebrovascular
development. Additionally, high Aβ1-40 exposure is linked to abnormal blood vessel formation and
cell death in zebrafish embryos. Beyond vascular changes, zebrafish models have been developed to investigate tauopathy,
a hallmark of AD. These models express mutant human tau proteins, leading to cytoskeletal disruptions and the
formation of NFTs similar to those found in human AD brains
[155-158]. In a groundbreaking
study, researchers successfully overexpressed human tau protein in zebrafish by replacing a native zebrafish
gene with human tau cDNA. This genetic
manipulation resulted in an eightfold increase in tau levels, leading to the accumulation of tau protein
within axons, mimicking the formation of NFTs observed in AD [159]. Recent
research has expanded the utility of zebrafish as an AD model. By introducing okadaic acid, a protein phosphatase
2A inhibitor, researchers have developed a pharmacological model that mimics AD-like pathologies. This treatment
leads to increased tau phosphorylation, similar to that observed in AD patients, and results in cognitive
and behavioral impairments [160]. Additionally, the formation of Aβ plaques is
enhanced in these zebrafish. Another model involves exposure to aluminum in acidic conditions, which
induces AD-like symptoms, including impaired swimming and learning abilities. These studies collectively demonstrate the
versatility of zebrafish as a platform for investigating AD pathogenesis and for developing potential
therapeutic interventions [161].
Zebrafish model for Aβ toxicity
The human APP has co-orthologs, appa, and appb, which show extensive and overlapping
expression patterns during early zebrafish embryogenesis, starting around midgastrulation. While these genes are expressed in
various tissues, appb is uniquely present throughout the developing spinal cord at 24 hours
post-fertilization [162, 163], whereas
appa is present in ependymal cilia, having critical role in cerebrospinal fluid flow and brain
homeostasis [164]. Lee and Cole linked a segment of the appb promoter to
green fluorescent protein in Danio rerio, revealing unexpected appb expression in the
emerging vasculature, contrary to previous observations. Similarly, appa and APP-like protein 2, which
contain transposon gene trap insertions encoding the red fluorescent protein, result in fusion proteins that
accumulate in the vasculature. Despite this, transcripts of these genes were not detected in endothelial cells but were
found in neurons, suggesting that the proteins are initially produced in neural cells before accumulating in the
vasculature [165]. To explore the function of appa and
appb, translation-blocking morpholinos were used [166].
Inhibiting appb led to abnormal cellular movements and reduced body length while suppressing appa had
no noticeable impact on embryonic development. Notably, injecting human APP mRNA rescued the appb-deficient
phenotype, although the FAD variant of human APP was less effective. Further research linked
appb deficiency to impaired axonal outgrowth and synapse formation during brain
development [167]. Song and Pimplikar demonstrated that both the extracellular and intracellular
domains of human APP are crucial for its function, as only full-length, not truncated, human APP could correct
neural defects. Hence these findings collectively highlight the zebrafish embryo as a valuable model for studying
various mutant forms of human APP [168]. Additional research has
indicated that the zebrafish psen1 wild type contributes to the abnormal production of Aβ1-42, a process
linked to familial FAD mutations [169]. Inhibiting psen1 translation
in zebrafish embryos through the use of morpholinos results in viable embryos but triggers p53-dependent neuronal
cell death. Furthermore, studies on psen1-deficient zebrafish mutants have unveiled a novel role
for psen1 in regulating the development of histaminergic neurons [170,
171]. These viable mutant fish provide evidence that psen1 is
essential for the growth of these specific neurons.
Zebrafish are a valuable model organism for studying the effects of hypoxia on various biological processes.
Researchers can induce hypoxic conditions in both adult and embryonic zebrafish using chemical methods, such as
administering sodium azide, or by directly reducing the oxygen levels in their aquatic environment
[163]. Importantly, hypoxia triggers the upregulation of appa,
bace1, appb, psen2, and psen1 genes in the brains of both adult and larval
zebrafish, mirroring the response observed in humans. These findings suggest that Aβ production in both humans and
zebrafish may serve as a protective mechanism against hypoxic conditions [172].
Transgenic zebrafish model for AD
Transgenic zebrafish, which are engineered to carry exogenous genes, offer a versatile platform for studying gene function, regulation, and overexpression, among other applications. Three main techniques i.e. meganuclease injection, transposon insertion, and microinjection or electroporation of linearized DNA, facilitate the creation of transgenic zebrafish embryos [161]. A commonly used method involves microinjecting embryos with a construct containing a transposon and transposase mRNA, leading to the integration of extrachromosomal DNA. This approach has been employed to create transgenic zebrafish expressing human MAPT to explore its function. Notably, zebrafish CNS neurons uniquely exhibit human MAPT expression. Bai et al. utilized the enolase 2 gene promoter to achieve high-level MAPT 4R expression in zebrafish neurons, resulting in tau protein accumulations that resemble NFTs [159]. Further developments by Paquet et al. used the HuC promoter to drive Gal4:VP16 expression in neurons, followed by the induction of DsRed and mutant TAU-P301L via a bidirectional promoter. These transgenic zebrafish showed biochemical alterations typical of human tauopathies. Additional models expressing Tau-P301L and Tau-A152T mutations were developed using the HuC promoter to closely mimic clinical features. These models display hyperphosphorylation, behavioral deficits, neuronal loss, and protein aggregation, similar to human tauopathies [170]. Lopez et al. observed reduced proteasome activity, increased tau phosphorylation, and neurodegeneration in Tau-A152T transgenic zebrafish, with autophagy promoting tau clearance [173]. Similarly, Tau-P301L-Tg zebrafish mainly showed tau hyperphosphorylation without significant oligomerization or NFT formation [174].
Zebrafish AD models for screening of new therapeutic agents
Zebrafish models have become essential for studying AD and evaluating potential treatments. Additionally, research has explored the therapeutic benefits of various compounds in zebrafish AD models. In a research AlCl3-induced zebrafish model linarin significantly reduced AChE activity and improved dyskinesia recovery [175]. Similarly, Necrostatin-1 was tested in an AlCl3-induced AD model, showing the potential to reverse learning and memory deficits, increase acetylcholine levels, and affect the expression of genes associated with necroptosis [47]. Furthermore, Silibinin and naringenin were found to alleviate neuroinflammation, oxidative stress, and neuroapoptosis in a bisphenol AlCl3-induced AD model, leading to improved cognitive function [176]. Another study examined the neuroprotective effects of TDZD-8 in an okadaic acid-induced AD model, showing that it decreased mortality, improved cognitive impairments, restored protein phosphatase 2A activity, and balanced GSK-3β phosphorylation [177].
Conclusions
AD is a multifaceted condition influenced by factors such as inflammation, oxidative stress, head injuries, genetics, and diabetes. While mammalian models have been used to study AD, their long lifespans, ethical concerns, and high costs presents significant challenges. To address these issues, researchers have turned to novel model organisms. This review highlights the potential of non-mammalian models, such as zebrafish, fruit flies, and worms, to advance our understanding of AD. These organisms offer advantages over traditional rodent models such as shorter lifespans, ethical permissibility, and lower maintenance costs. With advanced genetic tools, scientists can manipulate genes in these models to investigate their roles in AD development and progression Additionally, these models are crucial for screening environmental factors that might increase AD risk and for validating genetic risk factors identified in human studies. By leveraging these unique features, researchers can investigate the underlying mechanisms of AD, identify novel drug targets, and rapidly screen potential therapeutic compounds. As our knowledge of AD advances, these non-vertebrate models will remain essential for accelerating drug discovery and therapeutic development. While these models have proven to be valuable tools, it is important to recognize their limitations and to integrate them with other approaches, including human clinical studies and advanced imaging techniques. By combining the strengths of diverse model systems, researchers can gain a more comprehensive understanding of AD and ultimately develop effective strategies for its prevention and treatment.
Declarations
Acknowledgements
Not applicable.
Author's contribution
All the authors participated in the process of the initial writing of the manuscript, presentation of the idea, initial literature search, and review of the manuscript.
Availability of data and materials
Not Applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and informed consent
Not applicable.
References
1. Tiwari MK, & Kepp KP. β-amyloid pathogenesis: chemical properties versus cellular levels. Alzheimers Dement, 2016, 12(2): 184-194. [Crossref]
2. 2024 Alzheimer's disease facts and figures. Alzheimers Dement, 2024, 20(5): 3708-3821. [Crossref]
3. Gustavsson A, Norton N, Fast T, Frölich L, Georges J, Holzapfel D, et al. Global estimates on the number of persons across the Alzheimer's disease continuum. Alzheimers Dement, 2023, 19(2): 658-670. [Crossref]
4. Global, regional, and national burden of disorders affecting the nervous system, 1990-2021: a systematic analysis for the global burden of disease study 2021. Lancet Neurol, 2024, 23(4): 344-381. [Crossref]
5. Albadrani HM, Chauhan P, Ashique S, Babu MA, Iqbal D, Almutary AG, et al. Mechanistic insights into the potential role of dietary polyphenols and their nanoformulation in the management of Alzheimer's disease. Biomed Pharmacother, 2024, 174: 116376. [Crossref]
6. Chauhan P, Wadhwa K, Mishra R, Gupta S, Ahmad F, Kamal M, et al. Investigating the potential therapeutic mechanisms of puerarin in neurological diseases. Mol Neurobiol, 2024, 61(12): 10747-10769. [Crossref]
7. Ghofrani S, Joghataei MT, Mohseni S, Baluchnejadmojarad T, Bagheri M, Khamse S, et al. Naringenin improves learning and memory in an Alzheimer's disease rat model: insights into the underlying mechanisms. Eur J Pharmacol, 2015, 764: 195-201. [Crossref]
8. Hauptmann S, Scherping I, Dröse S, Brandt U, Schulz KL, Jendrach M, et al. Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging, 2009, 30(10): 1574-1586. [Crossref]
9. Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol, 2000, 1(2): 120-129. [Crossref]
10. Uwishema O, Mahmoud A, Sun J, Correia IFS, Bejjani N, Alwan M, et al. Is Alzheimer's disease an infectious neurological disease? A review of the literature. Brain Behav, 2022, 12(8): e2728. [Crossref]
11. Godoy JA, Rios JA, Zolezzi JM, Braidy N, & Inestrosa NC. Signaling pathway cross talk in Alzheimer's disease. Cell Commun Signal, 2014, 12: 23-33. [Crossref]
12. Firdaus Z, & Li X. Unraveling the genetic landscape of neurological disorders: insights into pathogenesis, techniques for variant identification, and therapeutic approaches. Int J Mol Sci, 2024, 25(4): 2320-2330. [Crossref]
13. Mukherjee P, Roy S, Ghosh D, & Nandi SK. Role of animal models in biomedical research: a review. Lab Anim Res, 2022, 38(1): 18-28. [Crossref]
14. Barré-Sinoussi F, & Montagutelli X. Animal models are essential to biological research: issues and perspectives. Future Sci OA, 2015, 1(4): Fso63. [Crossref]
15. Vandamme TF. Use of rodents as models of human diseases. J Pharm Bioallied Sci, 2014, 6(1): 2-9. [Crossref]
16. Strange K. Drug discovery in fish, flies, and worms. Ilar j, 2016, 57(2): 133-143. [Crossref]
17. Kimble J, & Nüsslein-Volhard C. The great small organisms of developmental genetics: Caenorhabditis elegans and Drosophila melanogaster. Dev Biol, 2022, 485: 93- 122. [Crossref]
18. Chauhan P, Wadhwa K, & Singh G. Caenorhabditis elegans as a model system to evaluate neuroprotective potential of nano formulations. Frontiers in Nanotechnology, 2022, 4: 1018754. [Crossref]
19. Rani N, Alam MM, Jamal A, Bin Ghaffar U, & Parvez S. Caenorhabditis elegans: a transgenic model for studying age-associated neurodegenerative diseases. Ageing res rev, 2023, 91: 102036. [Crossref]
20. Caldero-Escudero E, Romero-Sanz S, & De la Fuente S. Using C. elegans as a model for neurodegenerative diseases: methodology and evaluation. Methods Cell Biol, 2024, 188: 1-34. [Crossref]
21. Wang Q, Zhu BT, & Lei P. Animal models of Alzheimer's disease: current strategies and new directions. Zool Res, 2024, 45(6): 1385-1407. [Crossref]
22. Raabe A. High serum S100B levels for trauma patients without head injuries. Neurosurgery, 2001, 49(6): 1491- 1492; author reply 1492-1493. [Crossref]
23. Luo Y. Long-lived worms and aging. Redox Rep, 2004, 9(2): 65-69. [Crossref]
24. Alvarez J, Alvarez-Illera P, Santo-Domingo J, Fonteriz RI, & Montero M. Modeling Alzheimer's disease in Caenorhabditis elegans. Biomedicines, 2022, 10(2): 288-298. [Crossref]
25. Giong HK, Subramanian M, Yu K, & Lee JS. Non-rodent genetic animal models for studying tauopathy: review of drosophila, zebrafish, and C. elegans models. Int J Mol Sci, 2021, 22(16): 8465-8475. [Crossref]
26. McGurk L, Berson A, & Bonini NM. Drosophila as an in vivo model for human neurodegenerative disease. Genetics, 2015, 201(2): 377-402. [Crossref]
27. Lu B, & Vogel H. Drosophila models of neurodegenerative diseases. Annu Rev Pathol, 2009, 4: 315-342. [Crossref]
28. Nitta Y, & Sugie A. Studies of neurodegenerative diseases using Drosophila and the development of novel approaches for their analysis. Fly (Austin), 2022, 16(1):275-298. [Crossref]
29. Patton EE, & Zon LI. The art and design of genetic screens: zebrafish. Nat Rev Genet, 2001, 2(12): 956-966. [Crossref]
30. Dubey A, Kumari M, Sahu VK, Mishra A, & Dash SL. Zebrafish as a fascinating animal model: a robust platform for in vivo screening for biomedical researches. Int J Agricultural Sci Vet Med, 2024. [Crossref]
31. Liu Y. Zebrafish as a model organism for studying pathologic mechanisms of neurodegenerative diseases and other neural disorders. Cell Mol Neurobiol, 2023, 43(6): 2603-2620. [Crossref]
32. Saleem S, & Kannan RR. Zebrafish: an emerging realtime model system to study Alzheimer's disease and neurospecific drug discovery. Cell Death Discov, 2018, 4: 45-55. [Crossref]
33. Shenoy A, Banerjee M, Upadhya A, Bagwe-Parab S, & Kaur G. The brilliance of the zebrafish model: perception on behavior and Alzheimer's disease. Front Behav Neurosci, 2022, 16: 861155. [Crossref]
34. Wang X, Zhang JB, He KJ, Wang F, & Liu CF. Advances of zebrafish in neurodegenerative disease: from models to drug discovery. Front Pharmacol, 2021, 12: 713963. [Crossref]
35. Nigon VM, & Félix MA. History of research on C. elegans and other free-living nematodes as model organisms. WormBook, 2017, 2017: 1-84. [Crossref]
36. Van Pelt KM, & Truttmann MC. Caenorhabditis elegans as a model system for studying aging-associated neurodegenerative diseases. Transl Med Aging, 2020, 4: 60-72. [Crossref]
37. Roussos A, Kitopoulou K, Borbolis F, & Palikaras K. Caenorhabditis elegans as a model system to study human neurodegenerative disorders. Biomolecules, 2023, 13(3): 478-488. [Crossref]
38. Brenner S. The genetics of Caenorhabditis elegans. Genetics, 1974, 77(1): 71-94. [Crossref]
39. Shaye DD, & Greenwald I. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One, 2011, 6(5): e20085. [Crossref]
40. Ewald CY, Raps DA, & Li C. APL-1, the Alzheimer's amyloid precursor protein in Caenorhabditis elegans, modulates multiple metabolic pathways throughout development. Genetics, 2012, 191(2): 493-507. [Crossref]
41. Wasco W, Gurubhagavatula S, Paradis MD, Romano DM, Sisodia SS, Hyman BT, et al. Isolation and characterization of APLP2 encoding a homologue of the Alzheimer's associated amyloid beta protein precursor. Nat Genet, 1993, 5(1): 95-100. [Crossref]
42. Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature, 1987, 325(6106): 733-736. [Crossref]
43. Daigle I, & Li C. apl-1, a Caenorhabditis elegans gene encoding a protein related to the human beta-amyloid protein precursor. Proc Natl Acad Sci USA, 1993, 90(24): 12045-12049. [Crossref]
44. Hornsten A, Lieberthal J, Fadia S, Malins R, Ha L, Xu X, et al. APL-1, a Caenorhabditis elegans protein related to the human beta-amyloid precursor protein, is essential for viability. Proc Natl Acad Sci USA, 2007, 104(6): 1971- 1976. [Crossref]
45. Kim W, Underwood RS, Greenwald I, & Shaye DD. OrthoList 2: s new comparative genomic snalysis of human and Caenorhabditis elegans genes. Genetics, 2018, 210(2): 445-461. [Crossref]
46. Levitan D, & Greenwald I. Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer's disease gene. Nature, 1995, 377(6547): 351- 354. [Crossref]
47. Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature, 1995, 376(6543): 775-778. [Crossref]
48. Newman AP, White JG, & Sternberg PW. The Caenorhabditis elegans lin-12 gene mediates induction of ventral uterine specialization by the anchor cell. Development, 1995, 121(2): 263-271. [Crossref]
49. Li X, & Greenwald I. Membrane topology of the C. elegans SEL-12 presenilin. Neuron, 1996, 17(5): 1015-1021. [Crossref]
50. Levitan D, Doyle TG, Brousseau D, Lee MK, Thinakaran G, Slunt HH, et al. Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc Natl Acad Sci USA, 1996, 93(25): 14940-14944. [Crossref]
51. Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, et al. aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell, 2002, 3(1): 85-97. [Crossref]
52. L'Hernault SW, & Arduengo PM. Mutation of a putative sperm membrane protein in Caenorhabditis elegans prevents sperm differentiation but not its associated meiotic divisions. J Cell Biol, 1992, 119(1): 55-68. [Crossref]
53. Sarasija S, & Norman KR. Role of presenilin in mitochondrial oxidative stress and neurodegeneration in Caenorhabditis elegans. Antioxidants, 2018, 7(9): 111-121. [Crossref]
54. Cohen E, Bieschke J, Perciavalle RM, Kelly JW, & Dillin A. Opposing activities protect against age-onset proteotoxicity. Science, 2006, 313(5793): 1604-1610. [Crossref]
55. McColl G, Roberts BR, Gunn AP, Perez KA, Tew DJ, Masters CL, et al. The Caenorhabditis elegans a beta 1-42 model of Alzheimer disease predominantly expresses A beta 3-42. J Biol Chem, 2009, 284(34): 22697-22702. [Crossref]
56. Glenner GG, & Wong CW. Alzheimer's disease and down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun, 1984, 122(3): 1131-1135. [Crossref]
57. Bieschke J, Cohen E, Murray A, Dillin A, & Kelly JW. A kinetic assessment of the C. elegans amyloid disaggregation activity enables uncoupling of disassembly and proteolysis. Protein Sci, 2009, 18(11): 2231-2241. [Crossref]
58. Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, et al. Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron, 2008, 59(1): 43-55. [Crossref]
59. McColl G, Roberts BR, Pukala TL, Kenche VB, Roberts CM, Link CD, et al. Utility of an improved model of amyloidbeta (Aβ1-42) toxicity in Caenorhabditis elegans for drug screening for Alzheimer's disease. Mol Neurodegener, 2012, 7: 57-67. [Crossref]
60. Dimitriadi M, & Hart AC. Neurodegenerative disorders: insights from the nematode Caenorhabditis elegans. Neurobiol Dis, 2010, 40(1): 4-11. [Crossref]
61. Alexander AG, Marfil V, & Li C. Use of Caenorhabditis elegans as a model to study Alzheimer's disease and other neurodegenerative diseases. Front Genet, 2014, 5: 279- 289. [Crossref]
62. Fonte V, Kapulkin WJ, Taft A, Fluet A, Friedman D, & Link CD. Interaction of intracellular beta amyloid peptide with chaperone proteins. Proc Natl Acad Sci USA, 2002, 99(14): 9439-9444. [Crossref]
63. Fonte V, Kipp DR, Yerg J, 3rd, Merin D, Forrestal M, Wagner E, et al. Suppression of in vivo beta-amyloid peptide toxicity by overexpression of the HSP-16.2 small chaperone protein. J Biol Chem, 2008, 283(2): 784-791. [Crossref]
64. Perez N, Sugar J, Charya S, Johnson G, Merril C, Bierer L, et al. Increased synthesis and accumulation of heat shock 70 proteins in Alzheimer's disease. Brain Res Mol Brain Res, 1991, 11(3-4): 249-254. [Crossref]
65. Liang JJ. Interaction between beta-amyloid and lens alphaB-crystallin. FEBS Lett, 2000, 484(2): 98-101. [Crossref]
66. Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron, 2004, 43(3): 333-344. [Crossref]
67. Yochem J, Tuck S, Greenwald I, & Han M. A gp330/megalin-related protein is required in the major epidermis of Caenorhabditis elegans for completion of molting. Development, 1999, 126(3): 597-606. [Crossref]
68. McDermott JB, Aamodt S, & Aamodt E. ptl-1, a Caenorhabditis elegans gene whose products are homologous to the tau microtubule-associated proteins. Biochemistry, 1996, 35(29): 9415-9423. [Crossref]
69. Goedert M, Baur CP, Ahringer J, Jakes R, Hasegawa M, Spillantini MG, et al. PTL-1, a microtubule-associated protein with tau-like repeats from the nematode Caenorhabditis elegans. J Cell Sci, 1996, 109 (Pt 11): 2661- 2672. [Crossref]
70. Gordon P, Hingula L, Krasny ML, Swienckowski JL, Pokrywka NJ, & Raley-Susman KM. The invertebrate microtubule-associated protein PTL-1 functions in mechanosensation and development in Caenorhabditis elegans. Dev Genes Evol, 2008, 218(10): 541-551. [Crossref]
71. Treusch S, Hamamichi S, Goodman JL, Matlack KE, Chung CY, Baru V, et al. Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer's disease risk factors in yeast. Science, 2011, 334(6060): 1241-1245. [Crossref]
72. Link CD. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci USA, 1995, 92(20): 9368-9372. [Crossref]
73. Diomede L, Soria C, Romeo M, Giorgetti S, Marchese L, Mangione PP, et al. C. elegans expressing human β2-microglobulin: a novel model for studying the relationship between the molecular assembly and the toxic phenotype. PLoS One, 2012, 7(12): e52314. [Crossref]
74. He Q, Huang G, Chen Y, Wang X, Huang Z, & Chen Z. The protection of novel 2-arylethenylquinoline derivatives against impairment of associative learning memory induced by neural Aβ in C. elegans Alzheimer's disease model. Neurochem Res, 2017, 42(11): 3061-3072. [Crossref]
75. Fong S, Teo E, Ng LF, Chen CB, Lakshmanan LN, Tsoi SY, et al. Energy crisis precedes global metabolic failure in a novel Caenorhabditis elegans Alzheimer disease model. Sci Rep, 2016, 6: 33781. [Crossref]
76. Navarro-Hortal MD, Romero-Márquez JM, Osta S, Jimé- nez-Trigo V, Muñoz-Ollero P, & Varela-López A. Natural bioactive products and Alzheimer's disease pathology: lessons from Caenorhabditis elegans transgenic models. Diseases, 2022, 10(2): 28-38. [Crossref]
77. Teo E, Lim SYJ, Fong S, Larbi A, Wright GD, Tolwinski N, et al. A high throughput drug screening paradigm using transgenic Caenorhabditis elegans model of Alzheimer's disease. Transl Med Aging, 2020, 4: 11-21. [Crossref]
78. Brandt R, Gergou A, Wacker I, Fath T, & Hutter H. A Caenorhabditis elegans model of tau hyperphosphorylation: induction of developmental defects by transgenic overexpression of Alzheimer's disease-like modified tau. Neurobiol Aging, 2009, 30(1): 22-33. [Crossref]
79. Chen X, Bahramimehr F, Shahhamzehei N, Fu H, Lin S, Wang H, et al. Anti-aging effects of medicinal plants and their rapid screening using the nematode Caenorhabditis elegans. Phytomedicine, 2024, 129: 155665. [Crossref]
80. Paul D, Chipurupalli S, Justin A, Raja K, & Mohankumar SK. Caenorhabditis elegans as a possible model to screen anti-Alzheimer's therapeutics. J Pharmacol Toxicol Methods, 2020, 106: 106932. [Crossref]
81. McCormick AV, Wheeler JM, Guthrie CR, Liachko NF, & Kraemer BC. Dopamine D2 receptor antagonism suppresses tau aggregation and neurotoxicity. Biol Psychiatry, 2013, 73(5): 464-471. [Crossref]
82. Li H, Yu X, Li C, Ma L, Zhao Z, Guan S, et al. Caffeic acid protects against Aβ toxicity and prolongs lifespan in Caenorhabditis elegans models. Food Funct, 2021, 12(3): 1219-1231. [Crossref]
83. Xie Z, Zhao J, Wang H, Jiang Y, Yang Q, Fu Y, et al. Magnolol alleviates Alzheimer's disease-like pathology in transgenic C. elegans by promoting microglia phagocytosis and the degradation of beta-amyloid through activation of PPAR-γ. Biomed Pharmacother, 2020, 124: 109886. [Crossref]
84. Zhao L, Duan Z, Wang Y, Wang M, Liu Y, Wang X, et al. Protective effect of Terminalia chebula Retz. extract against Aβ aggregation and Aβ-induced toxicity in Caenorhabditis elegans. J Ethnopharmacol, 2021, 268: 113640. [Crossref]
85. Lu X, Zhang Y, Li H, Jin Y, Zhao L, & Wang X. Nicotine prevents in vivo Aβ toxicity in Caenorhabditis elegans via SKN-1. Neurosci Lett, 2021, 761: 136114. [Crossref]
86. Wu Y, Wu Z, Butko P, Christen Y, Lambert MP, Klein WL, et al. Amyloid-beta-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J Neurosci, 2006, 26(50): 13102-13113. [Crossref]
87. Diomede L, Rigacci S, Romeo M, Stefani M, & Salmona M. Oleuropein aglycone protects transgenic C. elegans strains expressing Aβ42 by reducing plaque load and motor deficit. PLoS One, 2013, 8(3): e58893. [Crossref]
88. Bai X, Liu CM, Li HJ, Zhang ZP, Cui WB, An FL, et al. Ethyl caffeate attefnuates Aβ-induced toxicity in Caenorhabditis elegans AD models via the insulin/insulin-like growth factor-1 signaling pathway. Bioorg Chem, 2023, 139: 106714. [Crossref]
89. Prüßing K, Voigt A, & Schulz JB. Drosophila melanogaster as a model organism for Alzheimer's disease. Mol Neurodegener, 2013, 8: 35-45. [Crossref]
90. Tsintzas E, & Niccoli T. Using Drosophila amyloid toxicity models to study Alzheimer's disease. Ann Hum Genet, 2024, 88(5): 349-363. [Crossref]
91. Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, et al. The genome sequence of Drosophila melanogaster. Science, 2000, 287(5461): 2185-2195. [Crossref]
92. Nichols CD. Drosophila melanogaster neurobiology, neuropharmacology, and how the fly can inform central nervous system drug discovery. Pharmacol Ther, 2006, 112(3): 677-700. [Crossref]
93. McGuire SE, Deshazer M, & Davis RL. Thirty years of olfactory learning and memory research in Drosophila melanogaster. Prog Neurobiol, 2005, 76(5): 328-347. [Crossref]
94. Bonner JM, & Boulianne GL. Drosophila as a model to study age-related neurodegenerative disorders: Alzheimer's disease. Exp Gerontol, 2011, 46(5): 335-339. [Crossref]
95. Cowan CM, Shepherd D, & Mudher A. Insights from Drosophila models of Alzheimer's disease. Biochem Soc Trans, 2010, 38(4): 988-992. [Crossref]
96. Rubin GM, Yandell MD, Wortman JR, Gabor Miklos GL, Nelson CR, Hariharan IK, et al. Comparative genomics of the eukaryotes. Science, 2000, 287(5461): 2204-2215. [Crossref]
97. Yagi Y, Tomita S, Nakamura M, & Suzuki T. Overexpression of human amyloid precursor protein in Drosophila. Mol Cell Biol Res Commun, 2000, 4(1): 43-49. [Crossref]
98. Fortini ME, Skupski MP, Boguski MS, & Hariharan IK. A survey of human disease gene counterparts in the Drosophila genome. J Cell Biol, 2000, 150(2): F23-30. [Crossref]
99. Luo L, Tully T, & White K. Human amyloid precursor protein ameliorates behavioral deficit of flies deleted for Appl gene. Neuron, 1992, 9(4): 595-605. [Crossref]
100.Carmine-Simmen K, Proctor T, Tschäpe J, Poeck B, Triphan T, Strauss R, et al. Neurotoxic effects induced by the Drosophila amyloid-beta peptide suggest a conserved toxic function. Neurobiol Dis, 2009, 33(2): 274-281. [Crossref]
101.Saad Y, Segal D, & Ayali A. Enhanced neurite outgrowth and branching precede increased amyloid-β-induced neuronal apoptosis in a novel Alzheimer's disease model. J Alzheimers Dis, 2015, 43(3): 993-1006. [Crossref]
102.Malmanche N, Dourlen P, Gistelinck M, Demiautte F, Link N, Dupont C, et al. Developmental expression of 4-repeat-tau induces neuronal aneuploidy in Drosophila tauopathy models. Sci Rep, 2017, 7: 40764. [Crossref]
103.Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, et al. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science, 2001, 293(5530): 711-714. [Crossref]
104.Folwell J, Cowan CM, Ubhi KK, Shiabh H, Newman TA, Shepherd D, et al. Abeta exacerbates the neuronal dysfunction caused by human tau expression in a Drosophila model of Alzheimer's disease. Exp Neurol, 2010, 223(2): 401-409. [Crossref]
105.Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, et al. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron, 2002, 34(4): 509-519. [Crossref]
106.Chatterjee S, Sang TK, Lawless GM, & Jackson GR. Dissociation of tau toxicity and phosphorylation: role of GSK- 3beta, MARK and Cdk5 in a Drosophila model. Hum Mol Genet, 2009, 18(1): 164-177. [Crossref]
107.Iijima-Ando K, Zhao L, Gatt A, Shenton C, & Iijima K. A DNA damage-activated checkpoint kinase phosphorylates tau and enhances tau-induced neurodegeneration. Hum Mol Genet, 2010, 19(10): 1930-1938. [Crossref]
108.Steinhilb ML, Dias-Santagata D, Mulkearns EE, Shulman JM, Biernat J, Mandelkow EM, et al. S/P and T/P phosphorylation is critical for tau neurotoxicity in Drosophila. J Neurosci Res, 2007, 85(6): 1271-1278. [Crossref]
109.Hua H, Münter L, Harmeier A, Georgiev O, Multhaup G, & Schaffner W. Toxicity of Alzheimer's disease-associated Aβ peptide is ameliorated in a Drosophila model by tight control of zinc and copper availability. Biol Chem, 2011, 392(10): 919-926. [Crossref]
110.Wu C, Wang J, Luo X, Wang B, Zhang X, Song Y, et al. Lead exposure induced transgenerational developmental neurotoxicity by altering genome methylation in Drosophila melanogaster. Ecotoxicol Environ Saf, 2024, 271: 115991. [Crossref]
111.Oliveira CS, Nogara PA, Lima LS, Galiciolli ME, Souza JV, Aschner M, et al. Toxic metals that interact with thiol groups and alteration in insect behavior. Curr Opin Insect Sci, 2022, 52: 100923. [Crossref]
112.Shilpa O, Anupama KP, Antony A, & Gurushankara HP. Lead (Pb)-induced oxidative stress mediates sex-specific autistic-like behaviour in Drosophila melanogaster. Mol Neurobiol, 2021, 58(12): 6378-6393. [Crossref]
113.Hu Y, Wu H, Lu C, Xu H, Li B, Guan W, et al. Cadmium chloride exposure impairs the growth and behavior of Drosophila via ferroptosis. Sci Total Environ, 2023, 865: 161183. [Crossref]
114.Wu Z, Du Y, Xue H, Wu Y, & Zhou B. Aluminum induces neurodegeneration and its toxicity arises from increased iron accumulation and reactive oxygen species (ROS) production. Neurobiol Aging, 2012, 33(1): 199.e191-112. [Crossref]
115.Liu B, Moloney A, Meehan S, Morris K, Thomas SE, Serpell LC, et al. Iron promotes the toxicity of amyloid beta peptide by impeding its ordered aggregation. J Biol Chem, 2011, 286(6): 4248-4256. [Crossref]
116.Abdulazeez R, Highab SM, Onyawole UF, Jeje MT, Musa H, Shehu DM, et al. Co-administration of resveratrol rescued lead-induced toxicity in Drosophila melanogaster. Environ Toxicol Pharmacol, 2024, 109: 104470. [Crossref]
117.Asejeje FO, Ogunro OB, Asejeje GI, Adewumi OS, & Abolaji AO. An assessment of the ameliorative role of hesperidin in Drosophila melanogaster model of cadmium chloride-induced toxicity. Comp Biochem Physiol C Toxicol Pharmacol, 2023, 263: 109500. [Crossref]
118.Fasae KD, Adeyemi O, Faleke HO, & Abolaji AO (2023). Chapter eleven-neurotoxicity of iron (Fe) in Drosophila and the protective roles of natural products. Advances in Neurotoxicology. J. Batista Rocha, M. Aschner and L. G. Costa, Academic Press. 9: 321-342.
119.Abolaji AO, Fasae KD, Iwezor CE, Aschner M, & Farombi EO. Curcumin attenuates copper-induced oxidative stress and neurotoxicity in Drosophila melanogaster. Toxicol Rep, 2020, 7: 261-268. [Crossref]
120.Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, Duthie FA, et al. Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer's disease. Neuroscience, 2005, 132(1): 123- 135. [Crossref]
121.Greeve I, Kretzschmar D, Tschäpe JA, Beyn A, Brellinger C, Schweizer M, et al. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J Neurosci, 2004, 24(16): 3899-3906. [Crossref]
122.Ye Y, & Fortini ME. Apoptotic activities of wild-type and Alzheimer's disease-related mutant presenilins in Drosophila melanogaster. J Cell Biol, 1999, 146(6): 1351- 1364. [Crossref]
123.Finelli A, Kelkar A, Song HJ, Yang H, & Konsolaki M. A model for studying Alzheimer's Abeta42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci, 2004, 26(3): 365-375. [Crossref]
124.Haddadi M, Nongthomba U, Jahromi SR, & Ramesh SR. Transgenic Drosophila model to study apolipoprotein E4-induced neurodegeneration. Behav Brain Res, 2016, 301: 10-18. [Crossref]
125.Wang L, Chiang HC, Wu W, Liang B, Xie Z, Yao X, et al. Epidermal growth factor receptor is a preferred target for treating amyloid-β-induced memory loss. Proc Natl Acad Sci USA, 2012, 109(41): 16743-16748. [Crossref]
126.Caesar I, Jonson M, Nilsson KP, Thor S, & Hammarström P. Curcumin promotes A-beta fibrillation and reduces neurotoxicity in transgenic Drosophila. PLoS One, 2012, 7(2): e31424. [Crossref]
127.Uras G, Manca A, Zhang P, Markus Z, Mack N, Allen S, et al. In vivo evaluation of a newly synthesized acetylcholinesterase inhibitor in a transgenic Drosophila model of Alzheimer's disease. Front Neurosci, 2021, 15: 691222. [Crossref]
128.Wang X, Perumalsamy H, Kwon HW, Na YE, & Ahn YJ. Effects and possible mechanisms of action of acacetin on the behavior and eye morphology of Drosophila models of Alzheimer's disease. Sci Rep, 2015, 5: 16127. [Crossref]
129.McKoy AF, Chen J, Schupbach T, & Hecht MH. A novel inhibitor of amyloid β (Aβ) peptide aggregation: from high throughput screening to efficacy in an animal model of Alzheimer disease. J Biol Chem, 2012, 287(46): 38992- 39000. [Crossref]
130.Kong Y, Li K, Fu T, Wan C, Zhang D, Song H, et al. Quercetin ameliorates Aβ toxicity in Drosophila AD model by modulating cell cycle-related protein expression. Oncotarget, 2016, 7(42): 67716-67731. [Crossref]
131.Sande R, Godad A, & Doshi G. Zebrafish experimental animal models for AD: a comprehensive review. Curr Rev Clin Exp Pharmacol, 2024, 19(4): 295-311. [Crossref]
132.Abou-Dahech MS, & Williams FE. Aging, age-related diseases, and the zebrafish model. J dementia Alzheimer's disease, 2024, 1(1): 48-71.
133.Chia K, Klingseisen A, Sieger D, & Priller J. Zebrafish as a model organism for neurodegenerative disease. Front Mol Neurosci, 2022, 15: 940484. [Crossref]
134.Lieschke GJ, & Currie PD. Animal models of human disease: zebrafish swim into view. Nat Rev Genet, 2007, 8(5): 353-367. [Crossref]
135.Nornes S, Newman M, Wells S, Verdile G, Martins RN, & Lardelli M. Independent and cooperative action of Psen2 with Psen1 in zebrafish embryos. Exp Cell Res, 2009, 315(16): 2791-2801. [Crossref]
136.Newman M, Nornes S, Martins RN, & Lardelli MT. Robust homeostasis of presenilin1 protein levels by transcript regulation. Neurosci Lett, 2012, 519(1): 14-19. [Crossref]
137.Musa A, Lehrach H, & Russo VA. Distinct expression patterns of two zebrafish homologues of the human APP gene during embryonic development. Dev Genes Evol, 2001, 211(11): 563-567. [Crossref]
138.Jiang H, Newman M, & Lardelli M. The zebrafish orthologue of familial Alzheimer's disease gene presenilin2 is required for normal adult melanotic skin pigmentation. PLoS One, 2018, 13(10): e0206155. [Crossref]
139.Groth C, Nornes S, McCarty R, Tamme R, & Lardelli M. Identification of a second presenilin gene in zebrafish with similarity to the human Alzheimer's disease gene presenilin2. Dev Genes Evol, 2002, 212(10): 486-490. [Crossref]
140.Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol, 2013, 31(3): 227- 229. [Crossref]
141.Moussavi Nik SH, Newman M, Ganesan S, Chen M, Martins R, Verdile G, et al. Hypoxia alters expression of zebrafish microtubule-associated protein tau (mapta, maptb) gene transcripts. BMC Res Notes, 2014, 7: 767- 777. [Crossref]
142.van Bebber F, Hruscha A, Willem M, Schmid B, & Haass C. Loss of Bace2 in zebrafish affects melanocyte migration and is distinct from Bace1 knock out phenotypes. J Neurochem, 2013, 127(4): 471-481. [Crossref]
143.Chen M, Martins RN, & Lardelli M. Complex splicing and neural expression of duplicated tau genes in zebrafish embryos. J Alzheimers Dis, 2009, 18(2): 305-317. [Crossref]
144.Mueller T, Vernier P, & Wullimann MF. The adult central nervous cholinergic system of a neurogenetic model animal, the zebrafish Danio rerio. Brain Res, 2004, 1011(2): 156-169. [Crossref]
145.Wullimann MF, Rupp B, & Reichert H (1996). The brain of the zebrafish Danio rerio: an overview.
146.Wullimann MF, Rupp B, & H. R (1996). The brain of the zebrafish Danio rerio: an overview. Neuroanat zebrafish brain. 1996:7-17.
147.Chen YC, Priyadarshini M, & Panula P. Complementary developmental expression of the two tyrosine hydroxylase transcripts in zebrafish. Histochem Cell Biol, 2009, 132(4): 375-381. [Crossref]
148.Anichtchik O, Sallinen V, Peitsaro N, & Panula P. Distinct structure and activity of monoamine oxidase in the brain of zebrafish (Danio rerio). J Comp Neurol, 2006, 498(5): 593-610. [Crossref]
149.Peri F, & Nüsslein-Volhard C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell, 2008, 133(5): 916- 927. [Crossref]
150.Kunst M, Laurell E, Mokayes N, Kramer A, Kubo F, Fernandes AM, et al. A cellular-resolution atlas of the larval zebrafish brain. Neuron, 2019, 103(1): 21-38.e25. [Crossref]
151.Low SE, Amburgey K, Horstick E, Linsley J, Sprague SM, Cui WW, et al. TRPM7 is required within zebrafish sensory neurons for the activation of touch-evoked escape behaviors. J Neurosci, 2011, 31(32): 11633-11644. [Crossref]
152.Nery LR, Eltz NS, Hackman C, Fonseca R, Altenhofen S, Guerra HN, et al. Brain intraventricular injection of amyloid-β in zebrafish embryo impairs cognition and increases tau phosphorylation, effects reversed by lithium. PLoS One, 2014, 9(9): e105862. [Crossref]
153.Best JD, Berghmans S, Hunt JJ, Clarke SC, Fleming A, Goldsmith P, et al. Non-associative learning in larval zebrafish. Neuropsychopharmacology, 2008, 33(5): 1206- 1215. [Crossref]
154.Cameron DJ, Galvin C, Alkam T, Sidhu H, Ellison J, Luna S, et al. Alzheimer's-related peptide amyloid-β plays a conserved role in angiogenesis. PLoS One, 2012, 7(7): e39598. [Crossref]
155.Donnini S, Solito R, Cetti E, Corti F, Giachetti A, Carra S, et al. Abeta peptides accelerate the senescence of endothelial cells in vitro and in vivo, impairing angiogenesis. Faseb j, 2010, 24(7): 2385-2395. [Crossref]
156.Luna S, Cameron DJ, & Ethell DW. Amyloid-β and APP deficiencies cause severe cerebrovascular defects: important work for an old villain. PLoS One, 2013, 8(9): e75052. [Crossref]
157.Bai Q, & Burton EA. Zebrafish models of tauopathy. Biochim Biophys Acta, 2011, 1812(3): 353-363. [Crossref]
158.Tomasiewicz HG, Flaherty DB, Soria JP, & Wood JG. Transgenic zebrafish model of neurodegeneration. J Neurosci Res, 2002, 70(6): 734-745. [Crossref]
159.Bai Q, Garver JA, Hukriede NA, & Burton EA. Generation of a transgenic zebrafish model of tauopathy using a novel promoter element derived from the zebrafish eno2 gene. Nucleic Acids Res, 2007, 35(19): 6501-6516. [Crossref]
160.Nada SE, Williams FE, & Shah ZA. Development of a novel and robust pharmacological model of okadaic acidinduced Alzheimer's disease in zebrafish. CNS Neurol Disord Drug Targets, 2016, 15(1): 86-94. [Crossref]
161.He X, Zhong ZM, & Che Y. Locomotor activity and learning and memory abilities in Alzheimer's disease induced by aluminum in an acid environment in zebrafish. Dongwuxue Yanjiu, 2012, 33(2): 231-236. [Crossref]
162.Banote RK, Chebli J, Şatır TM, Varshney GK, Camacho R, Ledin J, et al. Amyloid precursor protein-b facilitates cell adhesion during early development in zebrafish. Sci Rep, 2020, 10(1): 10127. [Crossref]
163.Moussavi Nik SH, Newman M, & Lardelli M. The response of HMGA1 to changes in oxygen availability is evolutionarily conserved. Exp Cell Res, 2011, 317(11): 1503-1512. [Crossref]
164.Chebli J, Rahmati M, Lashley T, Edeman B, Oldfors A, Zetterberg H, et al. The localization of amyloid precursor protein to ependymal cilia in vertebrates and its role in ciliogenesis and brain development in zebrafish. Sci Rep, 2021, 11(1): 19115. [Crossref]
165.Lee JA, & Cole GJ. Generation of transgenic zebrafish expressing green fluorescent protein under control of zebrafish amyloid precursor protein gene regulatory elements. Zebrafish, 2007, 4(4): 277-286. [Crossref]
166.Joshi P, Liang JO, DiMonte K, Sullivan J, & Pimplikar SW. Amyloid precursor protein is required for convergentextension movements during zebrafish development. Dev Biol, 2009, 335(1): 1-11. [Crossref]
167.Abramsson A, Kettunen P, Banote RK, Lott E, Li M, Arner A, et al. The zebrafish amyloid precursor protein-b is required for motor neuron guidance and synapse formation. Dev Biol, 2013, 381(2): 377-388. [Crossref]
168.Song P, & Pimplikar SW. Knockdown of amyloid precursor protein in zebrafish causes defects in motor axon outgrowth. PLoS One, 2012, 7(4): e34209. [Crossref]
169.Nornes S, Groth C, Camp E, Ey P, & Lardelli M. Developmental control of presenilin1 expression, endoproteolysis, and interaction in zebrafish embryos. Exp Cell Res, 2003, 289(1): 124-132. [Crossref]
170.Paquet D, Bhat R, Sydow A, Mandelkow EM, Berg S, Hellberg S, et al. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J Clin Invest, 2009, 119(5): 1382-1395. [Crossref]
171.Leimer U, Lun K, Romig H, Walter J, Grünberg J, Brand M, et al. Zebrafish (Danio rerio) presenilin promotes aberrant amyloid beta-peptide production and requires a critical aspartate residue for its function in amyloidogenesis. Biochemistry, 1999, 38(41): 13602-13609. [Crossref]
172.Moussavi Nik SH, Wilson L, Newman M, Croft K, Mori TA, Musgrave I, et al. The BACE1-PSEN-AβPP regulatory axis has an ancient role in response to low oxygen/oxidative stress. J Alzheimers Dis, 2012, 28(3): 515-530. [Crossref]
173.Lopez A, Lee SE, Wojta K, Ramos EM, Klein E, Chen J, et al. A152T tau allele causes neurodegeneration that can be ameliorated in a zebrafish model by autophagy induction. Brain, 2017, 140(4): 1128-1146. [Crossref]
174.Cosacak MI, Bhattarai P, Bocova L, Dzewas T, Mashkaryan V, Papadimitriou C, et al. Human tau(P301L) overexpression results in tau hyperphosphorylation without neurofibrillary tangles in adult zebrafish brain. Sci Rep, 2017, 7(1): 12959. [Crossref]
175.Pan H, Zhang J, Wang Y, Cui K, Cao Y, Wang L, et al. Linarin improves the dyskinesia recovery in Alzheimer's disease zebrafish by inhibiting the acetylcholinesterase activity. Life Sci, 2019, 222: 112-116. [Crossref]
176.Thayumanavan G, Jeyabalan S, Fuloria S, Sekar M, Ravi M, Selvaraj LK, et al. Silibinin and naringenin against bisphenol a-induced neurotoxicity in zebrafish modelpotential flavonoid molecules for new drug design, development, and therapy for neurological disorders. Molecules, 2022, 27(8). [Crossref]
177.Koehler D, Shah ZA, & Williams FE. The GSK3β inhibitor, TDZD-8, rescues cognition in a zebrafish model of okadaic acid-induced Alzheimer's disease. Neurochem Int, 2019, 122: 31-37. [Crossref]