Open Access | Research Article
This work is licensed under a Creative
Commons Attribution-ShareAlike 4.0 International License.
Additional polgD257A mutation (mutator) does not influence dopaminergic neurodegeneration in aged parkin-deficient mice
# These authors contributed equally to this work.
* Corresponding author: Xin-Ran Zhu
Mailing address: Department of Behavioral Neuroscience, RuhrUniversity Bochum, Universitätsstrasse 150, 44801 Bochum,
Germany.
Email: xinran.zhu@ruhr-uni-bochum.de
This article belongs to the Special Issue: Mitochondrial dysfunction in aging and aging-related diseases
Received: 25 December 2022 / Revised: 31 January 2023 / Accepted: 15 Feburary 2023 / Published: 29 March 2023
DOI: 10.31491/APT.2023.03.106
Abstract
Background: Parkinson’s disease is a neurodegenerative disease caused by the loss of dopaminergic neurons
in the substantia nigra pars compacta. Among the first identified causes of autosomal recessive Parkinson’s
disease were mutations in the parkin gene. Independently, we and other groups have developed various parkin
knockout mice, and none displayed dopaminergic degeneration in the substantia nigra. Interestingly, dopaminergic degeneration in the substantia nigra has been reported in a parkin knockout line (exon 3 deletion) carrying an additional mutation (D257A) in the mitochondrial DNA polymerase (polg) gene (mutator). The mutator mice show accelerated mutation rates in mitochondrial DNA resulting in a premature-aging phenotype.
Methods: To verify this finding, we crossed our parkin-deficient mice with the mutator mice, and characterized phenotypic changes of the parkin/mutator double mutant mice up to one year of age. We examined their
locomotion and motor coordination behaviors by using the open field, the rotarod, and the pole test, subsequently investigating their nigrostriatal axis by counting TH-positive cells in every tenth section throughout
the entire substantia nigra pas compacta and their termini in the striatum.
Results: The double mutants did not display additional deficiencies in locomotion in our behavior tests. We
could also not detect dopaminergic neurodegeneration in the substantia nigra pars compacta of aged double
mutants measured by levels of tyrosine hydroxylase positive neurons in the substantia nigra pars compacta as
well as in striatal terminals.
Conclusion: Our results do not support the hypothesis that the polgD257A mutation contributes to the age-related vulnerability of dopaminergic neurons in parkin-deficient mice.
Keywords
Parkin, neurodegeneration, polgD257A, mutator, substantia nigra
Introduction
Parkinson´s disease (PD) is characterized by a progressive
loss of dopaminergic (DA) neurons in the substantia nigra
pars compacta (SNc), leading to basal ganglia dysfunction
and subsequent impairment of movement control. To date
26 PD risk loci have been identified [1], providing substantial PD research advances via the generation of transgenic mouse models. One of the first genetic variations
found in PD patients was the exon 3 deletion in the parkin
gene, coding for an E3 ubiquitin ligase [2]. However,
all parkin knockout mouse strains did not show a hint of
loss of DA neurons in the SNc [3, 4]. The reason for the
missing neurodegeneration in those mouse models is not
understood. One could assume that the mouse DA neurons
in SNc are exposed to less aging since mice have a shorter
lifespan. This explanation is supported by findings that
the age-dependent mutation rate of mitochondrial DNA
(mtDNA) increases much more in human SNc compared
to murine tissue [5]. Such age-dependent accumulation of
mtDNA mutations, as well as a broad spectrum of aginglike phenotypes, have been observed in the polgD257A mice
(mutator; MT) by a knock-in proofreading-deficient version of the mtDNA polymerase [6]. Utilizing this mouse
model, Pickrell et al., 2015 reported age-dependent degeneration of DA neurons in the SNc in aged MT/Parkin
KO double mutants [7].
In this study, we intended to verify this finding by crossing MT mice with our parkin-deficient Padel mice [8].
We followed possible phenotypic changes of MT/Padel
double mutant mice up to one year of age, but could not
find any additional deficiency in locomotion, nor dopaminergic neurodegeneration in the SNc compared to MT and
wild-type mice. Our results do not support the hypothesis
that the polgD257A mutation contributes to the age-related
vulnerability of dopaminergic neurons in parkin-deficient
mice.
Materials and methods
Animals
All animal experiments were conducted and approved using the German guidelines and by the animal care and use
committee of the state of North-Rhine Westphalia. Mice
were kept under standard conditions with free access to
water and food and a 12:12 hrs a day/night cycle.
The parkin knockout Padel carries a deletion for the exon
3 in the parkin gene, and the neo gene was removed by
crossing with the Deleter followed by backcrossing with
C57BL/6N mice for over 20 generations [8]. The Bl6
congenic Mutator mice (MT) were acquired from the
Jackson Laboratory (B6.129S7(Cg)-Polgtm1Prol/J, Strain
#:017341). Animals of both sexes were used for all behavior and histological studies.
Behavior tests
The rotarod test was conducted on a rod starting with 4
rpm and accelerating to 40 rpm within 240 s and the latency to fall was measured (RotaRod, TSE, Germany).
For the pole test mice (12 months old) were placed on top
of a metal pole (height: 55 cm; diameter: 8 mm), and the
time until the animals reached the ground was measured.
Further, it was noted when the animals fell or slipped off
the pole.
For the open field tests, the mice were placed in a plastic
box (40 × 40 × 40 cm) for 30 min. Their traveled distance,
number of stops, time of rest, and the time spent in the
center of the box (20 × 20 cm) were recorded by the Videomot2 software (TSE, Germany).
Immunohistochemistry
Mice were perfused transcardially with 4% formaldehyde. Brains were removed and dehydrated with ethanol and isopropanol followed by embedding in paraffin. Coronal brain sections (10 µm) of the SN and striatum were made (RM2145, Leica, Germany). These sections were deparaffinized and rehydrated followed by incubation with the primary antibody (anti-TH; 1:1000; Merk Millipore; ab152). Subsequently, sections were incubated with the secondary antibody (biotinylated anti-rabbit IgG; 1:300; Vector Laboratories; BA-1000) followed by incubation with an avidin/biotin complex solution (vectastain® elite ABC-HRP kit; Vector Laboratories) and stained with diaminobenzidine.
Quantifications and statistical analysis
Every tenth consecutive section containing TH positive cells within the entire SN was counted using the software ImageJ. The staining intensity units of TH positive axonal terminals in the striatum were analyzed with CellProfilerTM (Module: MeasureImageIntensity). All statistical analyses with H- and u-tests were performed with the software SigmaPlot 14.0 (Systat Software).
Results
Four homozygous genotypes (wild type (WT) littermates
(LM): WT/WT; MT: polg-/-; Padel: Padel-/-; MT/Padel:
polg-/-/ Padel-/-) were used to examine phenotypic changes
up to an age of 12 months since the median lifespan of homozygous MT is 416 days [6]. We note that MT and MT/
Padel mice started to lose body weight when the animals
approached 8 months of age (Figure 1A), consistent with
published results [7]. In addition, we also observed spleen
enlargement (Figure 1B) and shorter lifespan (Figure 1C)
of MT/Padel mice, which was not different from that of
MT mice. Based on those parameters we confirmed the
premature aging phenotype of the MT/Padel mice.
Next, we investigated whether the combination of the
polgD257A mutation with the parkin deletion may contribute
to additional motor impairments that could indicate loss
of DA neurons in SNc. Six-month-old mice were tested
monthly, up to 12 months of age in the rotarod task. Consistent with our previous results [4], the Padel and WT
mice showed similar rotarod performance (Figure 1D).
While the MT mice showed low performance constantly,
the MT/Padel mice showed an age-dependent, continuous
decline in latency to fall (Figure 1D). However, there was
no difference in the rotarod performance between MT and
MT/Padel mice at the age of 7 to 12 months, indicating
that the motor impairment of the aged MT/Padel mice was
dependent only on the polgD257A mutation.
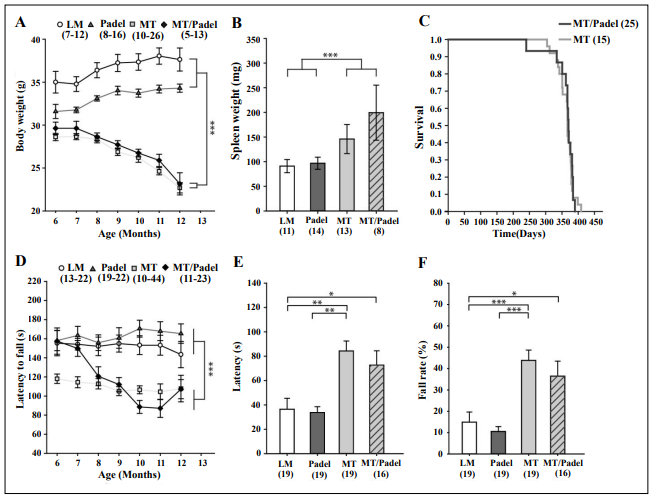
Figure 1. MT and MT/Padel mice displayed similar phenotypical effects. (A) Body weight (mean ± SEM) of Padel mice at the age of 6 months was lower than wildtype littermates (LM) (u-test: p < 0.05), increased after 8. Month of age (H-test: p < 0.05), which was then not different from that of LM (u-tests: p > 0.05). MT and MT/Padel mice started to lose body weight when the animals approached 8 months of age (H-tests: p < 0.001), which was different from LM and Padel (u-tests: p < 0.05). There were no differences between MT and MT/Padel mice (u-tests: p > 0.05). (B) MT and MT/ Padel mice (12 months old) showed similar enlargement of spleen compared to LM and Padel (mean ± SEM; u-test: p > 0.05 for MT vs MT/Padel). (C) MT and MT/Padel mice exhibited similar survival rates in Kaplan-Meier analysis (log-rank test: p > 0.05). (D) LM and Padel mice exhibited similar constant performance (mean ± SEM) on the rotarod (u-tests: p > 0.05). MT mice showed low performance, compared to LM and Padel (u-tests: p < 0.05). MT/Padel mice showed an age dependent, continuous decline in performances (H-test: p < 0.001). No difference was found between MT and MT/Padel for 8-12 months. (u-tests; p > 0.05). (E) LM and Padel mice displayed similar latencies (mean ± SEM) in the pole test (u-test: p > 0.05). MT and MT/Padel mice showed reduced performance compared to LM and Padel (u-tests: p < 0.05). No differences were found between MT and MT/Padel (u-test: p > 0.05). (F) MT and MT/Padel mice exhibited a higher percentage of trials in which they fell off or slid down the pole during the pole test (mean ± SEM). No difference was found between MT and MT/Padel mice (u-test; p > 0.05). (A-F) Numbers in parentheses indicate numbers of analysed animals. H- or u-test: *p < 0.05; **p < 0.01; ***p < 0.001.
Analogs to the rotarod task, mice were tested in the open field starting at the age of six months to examine locomotor activity and habituation behavior. Young WT mice exhibited locomotion habituation in the open field over time, showing reduced levels of activity on the second day compared to the first day, while our Padel mice displayed delayed habituation which was visible on the third day (Figure 2A, left). The same-aged MT and MT/Padel mice exhibited reduced locomotor activity on the first day. However, the delayed habituation behavior could be observed only in MT/Padel mice, but not in MT mice (Figure 2A, left). This result indicates that the delayed habituation was caused by the parkin deletion alone, while the polgD257A mutation was responsible for the reduced locomotor activity of the MT/Padel mice. A similar phenomenon was also observed in the second parameter of the open field: The time in the center. Interestingly, the MT/Padel mice seem to spend more time in the center region of the open field compared with the WT mice. However, this effect was dependent on the polgD257A mutation only (Figure 2A, right). While the WT and Padel mice exhibited relatively constant locomotor activity when they were re-exposed to the open field, two groups of these mice with the polgD257A mutation showed an age-dependent decline in the total traveled distance and increased resting time, regardless of the parkin deletion (Figure 2B). This polgD257A-dependent effect could also be observed in stops, a parameter reflecting the initiation of movement (Figure 2A + B).
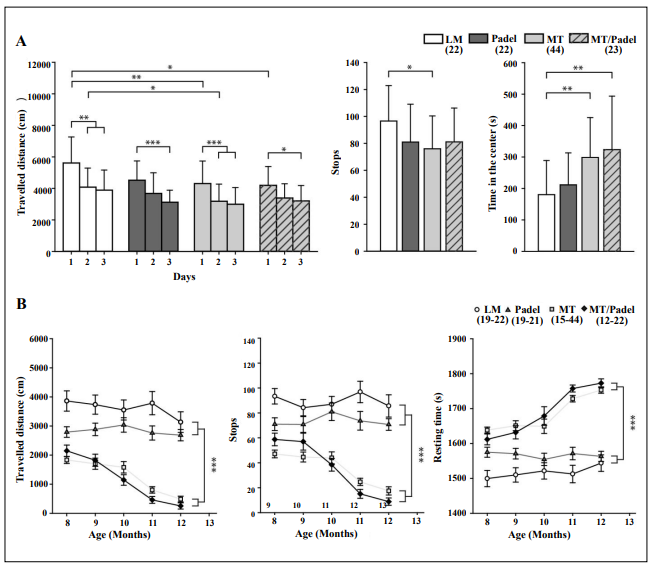
Figure 2. MT and MT /Padel mice exhibited similar phenotypical changes in the open field test. (A) Naïve mice (6–7 months old) were exposed to the open field for 30 min on three consecutive days. (Left) The MT and MT/Padel mice showed reduction of the travelled distance on the day 1, while no difference was found between LM and Padel mice (u-test: p > 0.05). LM and MT mice displayed similar habituation behaviour by showing reduced travelled distance on the day 2 and 3, while a delayed habituation on the day 3 of the test was found for Padel and MT/Padel animals. (Middle) Averaged number of stops within 30 min was lower with MT mice compared to LM. No difference was found between MT and MT/Padel (u-test: p > 0.05). (Right) MT and MT/Padel mice showed similarly increased amounts of time spend in the center compared to LM and Padel. No differences found between MT and MT/Padel (u-test: p > 0.05). (B) MT and MT/Padel mice showed similar successive shortening of travelled distances (left), similarly less initiative movements (as stops) (middle), and similar increased resting time (right), when the animals were re-tested monthly in the open field up to 12 months of age. (A-B) All data: mean ± SEM, Numbers in parentheses indicate numbers of analysed animals. H- or u-test: *p < 0.05; ** p< 0.01; ***p < 0.001.
So far, our behavior tests showed severe impairment of
MT/Padel double mutant mice in motor coordination and
locomotor activity. However, this impairment seems to
be caused by the polgD257A mutation alone, since there was
no difference between animals with or without the parkin
deletion. These results contradict the finding reported by
Pickrell et al., [7] using the pole test, an alternative behavior test for dopamine-dependent motor coordination.
To examine this discrepancy, we performed the pole test
using a similar protocol described by Pickrell et al., [7]
and found significantly higher latency time descending the
pole for MT/Padel double mutants compared with WT or
Padel mice (Figure 1E). However, MT mice displayed a
similar higher latency time that was statistically not different from those of MT/Padel double mutants (Figure 1E).
This polgD257A-dependent impairment was also detected
in the number of mice that fell off or slid down the pole
(Figure 1F). This result is consistent with those observed
in our rotarod and the open field test and demonstrates impairment of aged MT/Padel double mutant mice in locomotor activity and motor coordination, which is dependent on the polgD257A mutation only.
After the last behavioral test at the age of 12 months, mice
were sacrificed to determine the integrity of their nigrostriatal axis by counting TH-positive cells in every tenth
section throughout the entire SNc. Mice with all four genotypes displayed similar counts of TH-positive neurons in
their SNc (Figure 3A + C). To determine if there was any
change regarding the nigrostriatal axis in the MT/Padel
mice, we also counted the intensities of the TH-staining
in the striatum and compared them with Padel, MT, and
wild-type mice. We were not able to find any significant
difference in striatal TH staining intensity between all
four groups (Figure 3B + D). Our results demonstrate that
those behavioral impairments of the MT and MT/Padel
mice in locomotor activity and motor coordination were
likely not due to dopamine deficiency in their nigrostriatal
axis.
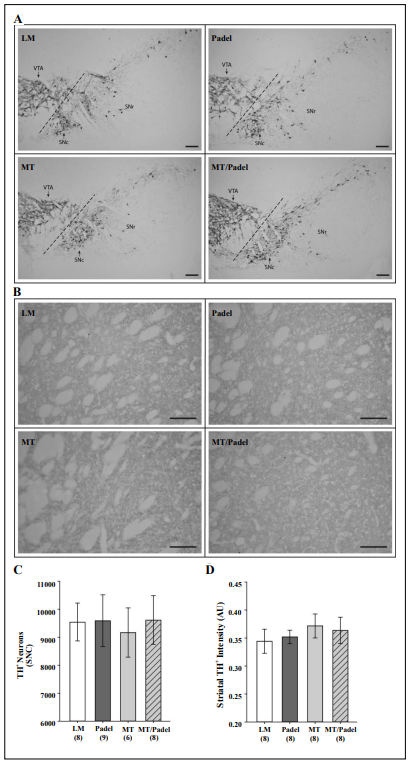
Figure 3. No significant morphological difference in nigrostriatal axis between wildtype (LM), Padel, MT and MT/Padel mice. (A) Representative images of TH-stained coronal brain sections containing the substantia nigra pars compacta (SNc), substantia nigra pars reticulata, (SNr) and ventral tegmental area (VTA) at the age of 12-months. ( B ) Representative images of TH-stained coronal brain sections containing the striatum for each of those four genotypes at the age of 12 months. (C) Similar TH positive cell counts (mean ± SD) in the SNc (H-test; p > 0.05) (D) Similar TH-staining intensity units (mean ±SD) in the striatum between four genetypes (H-Test; p > 0.05). (A-B) scale bars: 100 µm. (C-D) Numbers in parentheses i n d i c a t e n u m b e r s o f analysed animals.
Discussion
Pickrell et al., reported that the MT/Parkin KO mice exhibited age-dependent degeneration of DA neurons in the
SNc and motor deficit in the pole test [7]. By crossing MT
mice with our Padel mice we intended to verify this finding with an independent mouse strain carrying the same exon 3 deletions of the parkin gene. Some characteristic
features for premature aging, e.g. lost body weight, spleen
enlargement, and short lifespan could also be confirmed
in the MT/Padel mice, which were indistinguishable from
those of MT mice (Figure 1A - C). However, there are several discrepancies in motor behaviors between the results
of the pole test in both studies. We observed similar impairment of MT mice in latency time descending the pole
with or without the parkin deletion (Figure 1E), while the
MT mice did not exhibit such motor impairment in the
study of Pickrell et al., [7]. Because of their severe aging
phenotypes, it seems less likely that the aged MT mice are
still able to perform such complex motor tasks on a wildtype level. Indeed, a study by Hauser et al., demonstrated
that the old MT mice tend to slide down or fall off instead
of climb down the pole [9], which could be confirmed
in our study (Figure 1F). Additionally, we found that the
slide-down rates of MT (mean ± SEM: 28.33 ± 10.70 %)
and MT/Padel (mean ± SEM: 35.91 ± 5.39 %) mice were
significantly higher than those of wild type (mean ± SEM:
10.65 ± 4.22 %) and Padel (mean ± SEM: 5.26 ± 2.23 %)
mice (H- and u-test, p < 0.05). This higher slide-down rate
was not dependent on the parkin deletion (H- and u-test,
p > 0.05), but on the MT mutation. Furthermore, impairment in locomotion and motor coordination has also been
observed in two previous studies, when aged MT mice
were tested in the open field and on the rotarod [10, 11].
By their results, we observed similar motor impairments
for the aged MT/Padel mice in both tests, most likely independent of the parkin mutation.
Consistent with our behavior results, no significant difference was found in the number of the TH positive cell bodies in the SNc and their terminals in the striatum between
wild-type, Padel, MT, and MT/Padel mice in our study
(Figure 3). Those observed motor deficits in the aged MT
mice seem not to be related to functions of the nigrostriatal axis. This result is also supported by the fact that Ldopa could not restore the normal motor behavior of the
MT mice [9]. Altogether, we were not able to confirm the
protective function of parkin against the degeneration of
DA neurons in the SNc of the MT mice described by Pickrell et al., Although the Parkin KO strains used in both
studies share a similar deletion of the exon 3 in the parkin
gene, there are few differences in their genomic region
surrounding the deletion site. While a 34 base pair loxP
remains in this region in our Padel mouse, the Parkin KO
strain used in the study of Pickrell et al., carried the neoresistant gene with an additional GFP coding sequence [3].
We speculate that the additional big DNA fragment coding for two functional proteins under a strong promoter
may cause DA neurons more susceptible to degeneration
during aging in the MT background with deficient parkin
function. It is also known that the neo-resistant gene can
result in altered expression of other genes as much as 100-
kilo bases distant in the locus downstream of the pGK
NEO insertion since its excision from the locus results in
dramatically different phenotypes [12].
Our results are similar to other studies that have crossed
MT mice with other mouse models for PD. The double
mutant of MT with DJ-1 KO did not affect the numbers of
TH-positive cells in the SNc up to 12 months of age [9].
Likewise, treatment of MT mice with the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a toxin mouse
model for PD, did not result in an additional degeneration of DA neurons in the nigrostriatal axis [13]. With the
same MT/Parkin KO, parkin deficiency does not affect the
cardiac hypertrophy observed in aged MT mice [14]. Several studies have shown that aged MT mice carry mtDNA
deletions in the SNc to a similar extent as that found in
humans, but with no difference between PD patients and
controls [15]. Altogether, this suggests that the absence of
parkin was unlikely to have a strong effect on the survival
of SNc DA neurons in aged MT mice and contradicts results found by Pickrell et al., [7]. A difference in genetic
background around the exon 3 deletion site in both parkin
KO strains seems to be the most likely explanation for
these conflicting results. Therefore, at least two independent strains should be included in such investigations in
the future.
Conclusion
Our results do not support the hypothesis that the polgD257A mutation contributes to the age-related vulnerability of dopaminergic neurons in parkin-deficient mice.
Declarations
Availability of data and materials
The data that support the findings of this study are available from the corresponding author, Zhu XR., upon request.
Financial support and sponsorship
Mark DM. is supported by the German Research Foundation, MA 5806/2- 1.
Conflicts of interest
The author declares that there are no conflicts.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
References
1. Lill CM. Genetics of Parkinson’s disease. Mol Cell Probes, 2016, 30(6): 386-396. [Crossref]
2. Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet, 2000, 25(3): 302-305. [Crossref]
3. Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem, 2003, 278(44): 43628-43635. [Crossref]
4. Zhu XR, Maskri L, Herold C, Bader V, Stichel CC, Güntürkün O, et al. Non-motor behavioural impairments in parkin-deficient mice. Eur J Neurosci, 2007, 26(7): 1902-1911. [Crossref]
5. Guo X, Kudryavtseva E, Bodyak N, Nicholas A, Dombrovsky I, Yang D, et al. Mitochondrial DNA deletions in mice in men: substantia nigra is much less affected in the mouse. Biochim Biophys Acta, 2010, 1797(6-7): 1159- 1162. [Crossref]
6. Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science, 2005, 309(5733): 481-484. [Crossref]
7. Pickrell AM, Huang CH, Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, et al. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron, 2015, 87(2): 371-381. [Crossref]
8. Rybarski M, Mrohs D, Osenberg K, Hemmersbach M, Pfeffel K, Steinkamp J, et al. Loss of parkin causes endoplasmic reticulum calcium dyshomeostasis by upregulation of reticulocalbin 1. Eur J Neurosci, 2023. [Crossref]
9. Hauser DN, Primiani CT, Langston RG, Kumaran R, & Cookson MR. The Polg Mutator Phenotype Does Not Cause Dopaminergic Neurodegeneration in DJ-1-Deficient Mice. eNeuro, 2015, 2(1). [Crossref]
10. Dai Y, Kiselak T, Clark J, Clore E, Zheng K, Cheng A, et al. Behavioral and metabolic characterization of heterozygous and homozygous POLG mutator mice. Mitochondrion, 2013, 13(4): 282-291. [Crossref]
11. Ross JM, Coppotelli G, Branca RM, Kim KM, Lehtiö J, Sinclair DA, et al. Voluntary exercise normalizes the proteomic landscape in muscle and brain and improves the phenotype of progeroid mice. Aging Cell, 2019, 18(6): e13029. [Crossref]
12. Gingrich JA, & Hen R. The broken mouse: the role of development, plasticity and environment in the interpretation of phenotypic changes in knockout mice. Curr Opin Neurobiol, 2000, 10(1): 146-152. [Crossref]
13. Dai Y, Clark J, Zheng K, Kujoth GC, Prolla TA, & Simon DK. Somatic mitochondrial DNA mutations do not increase neuronal vulnerability to MPTP in young POLG mutator mice. Neurotoxicol Teratol, 2014, 46: 62-67. [Crossref]
14. Woodall BP, Orogo AM, Najor RH, Cortez MQ, Moreno ER, Wang H, et al. Parkin does not prevent accelerated cardiac aging in mitochondrial DNA mutator mice. JCI Insight, 2019, 5(10). [Crossref]
15. Perier C, Bender A, García-Arumí E, Melià MJ, Bové J, Laub C, et al. Accumulation of mitochondrial DNA deletions within dopaminergic neurons triggers neuroprotective mechanisms. Brain, 2013, 136(Pt 8): 2369-2378. [Crossref]