Open Access | Review
This work is licensed under a Creative
Commons Attribution-ShareAlike 4.0 International License.
Influence of metformin on age-related macular degeneration
* Corresponding author: Albert J. Augustin
Mailing address: Department of Ophthalmology, Staedtisches
Klinikum Karlsruhe, Moltkestr. 90, 76133 Karlsruhe, Germany.
Email: albertjaugustin@googlemail.com
This article belongs to the Special Issue: Enhancing healthy aging with drugs that target insulin signaling
Received: 26 August 2022 / Revised: 19 September 2022 / Accepted: 21 November 2022 / Published: 29 December 2022
DOI: 10.31491/APT.2022.12.098
Abstract
Metformin is the most commonly prescribed antihyperglycemic drug as first-line therapy in type II diabetic patients. In recent years, evidence is increasing that metformin has beneficial effects beyond its classical antihyperglycemic way of action. Those effects include anti-inflammation, anti-oxidation, anti-aging, anti-angiogenesis, anti-neoplasia, anti-apoptosis, and neuroprotection. The complex pathophysiology of age-related macular degeneration (AMD) includes age-related changes in the retinal pigment epithelium (RPE) and Bruch’s membrane. An inflammatory and oxidative damage component has also been described. The dry form of late AMD is especially characterized by degeneration of the RPE, Bruch’s membrane, the choriocapillaris and finally, loss of the photoreceptors (geographic atrophy), and the wet form of late AMD is characterized by pathological neovascularization. An increasing number of reports about the beneficial effects of metformin on AMD have been published in the last few years. Several effects of metformin could be linked to the AMPK pathway. A first prospective trial investigating the effect of metformin on dry AMD is ongoing with estimated results by the end of 2024. In this review, the current knowledge about the association between metformin and AMD is summarized.
Keywords
Metformin, age-related macular degeneration retina, insulin, diabetes, aging, drug therapy, AMPK pathway
Introduction
Metformin is one of the most commonly used oral antidiabetic drugs. Classically, it is used in non-insulindependent type 2 diabetic patients and most cases as the
first oral antidiabetic medication. Metformin inhibits the
formation of glucose in the liver and improves glucose
turnover in the periphery (the muscles) of the body, thereby lowering the blood glucose level [1, 2].
There is increasing evidence that metformin may exert
several beneficial effects beyond its original antidiabetic
function [3-5]. In summary, in vitro, and in vivo investigations report anti-angiogenic, anti-inflammatory, antioxidative, anti-apoptotic, anti-aging, and neuroprotective
effects of metformin [6, 7]. Most of these effects also play
a crucial role in many retinal diseases such as diabetic retinopathy (DR), age-related macular degeneration (AMD),
glaucoma, uveitis, or inherited retinal dystrophies as retinitis pigmentosa.
AMD is a vision-threatening disease of the elderly population worldwide with increasing prevalence. Wong et al.
calculated an increase from 196 million affected people in
2020 to 288 million affected people in 2040 [8]. Together
with diabetic retinopathy and glaucoma, AMD accounts
for the majority of legal blindness cases in developed
countries. In Germany, for example, it is estimated that
AMD is responsible for up to 50% of legally blind people
[9] .
AMD is a progressive, multi-factorial disease with a complex pathophysiology that is still not fully understood in
all its details. The main risk factor is age. It is also known
that a history of smoking, hyperlipidemia, ethnicity, and
a certain genetic disposition as well as inflammatory processes play a role [10, 11]. Clinically, AMD is classified
into different stages: an early, an intermediate, and two
late stages: dry, non-neovascular, and wet, neovascular
late-stage AMD (Figure 1) [12]. The early and intermediate stages are characterized by the size of the drusen
deposits and by the presence or absence of pigmentary
changes. The early and intermediate stages usually have
no or only minimal symptoms [10]. Late, neovascular
AMD, however, has more profound visual symptoms that
can progress rapidly. The symptoms include distortion
and/or large central scotoma or blind spot due to hemorrhage or fluid accumulation in the macular region. If left
untreated, fibrosis and permanent vision loss are the consequences [10]. Late, non-neovascular AMD is characterized by progressive central vision loss due to degeneration of the retinal pigment epithelium (RPE) and the photoreceptor cells, referred to as geographic atrophy (GA) [10, 11].
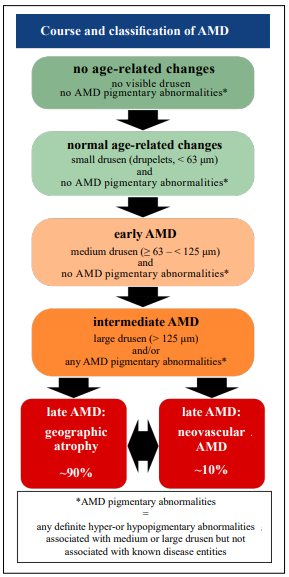
Figure 1. Classification and course of age-related macular degeneration (AMD). The earliest precursor signs of AMD are small drusen that are classified as normal age-related changes. Early AMD is characterized by the presence of medium drusen but the absence of AMD pigmentary changes which are defined as any hyper- or hypopigmentary abnormality. Intermediate AMD shows large drusen and/or the presence of any AMD pigmentary abnormalities. The late stages of AMD are its two distinct forms: neovascular AMD (wet AMD) and geographic atrophy (dry AMD) with the latter being the more common form. Both forms may merge into one another or be present simultaneously. (modified from Ferris et al. [12])
Approved treatment options are currently only available for late, neovascular AMD. The Standard of care is the intravitreal injection of anti-VEGF agents to block angiogenic factors that induce the formation of pathological neovascular vessels. Pathologic neovascularization leads to retinal damage by sub- and/or intraretinal fluid or blood accumulation [13]. Additionally, larger subretinal hemorrhages can be treated by surgical intervention to eliminate the subretinal blood mechanically [14]. The late, dry stage of AMD remains untreatable to date thus efforts are made to find a way to modify the disease. In the last years, an increasing number of scientific publications report on several potential associations of metformin with the course of the disease. This is true for both the development and the treatment of AMD. In this review, we summarize the current knowledge about these associations and the potential underlying (patho)physiological mechanisms.
Methods
The systematic literature search was performed using the
PubMed library. The search term “metformin age-related
macular degeneration” revealed a total of 35 publications
(the search was performed on July 20th, 2022). After the
screening of titles and abstracts, 22 publications qualified as being suited for the topic of this review. Further
database searches with adjusted search terms (metformin
AMD, metformin macular degeneration pathways, etc.)
did not reveal any further relevant articles.
Additional publications have been included for the introductory part as well as for the background part on the
pathophysiology of AMD and the mode of action of metformin. These publications were identified by direct database search as well as by backward citation searching.
Pathophysiology of AMD
As mentioned above, the pathophysiology of AMD is
complex and several risk factors are associated with this
disease. As a neurosensory tissue, the retina, especially
the photoreceptor cells, is metabolically highly active.
This requires a constant balance between the breakdown
of metabolic waste products and the supply of necessary
nutrients, including oxygen. In the healthy retina, the RPE
with its tight contact with the photoreceptor cells, Bruch’s
membrane, and the choroidal vasculature execute this important task [15]. The RPE cells form a single cell layer
with neighboring cells being connected via tight junctions.
This single-cell layer is supported by Bruch’s membrane
lying underneath. This complex forms the selective diffusion barrier known as the blood-retina-barrier (BRB)
which precisely regulates the passage of ions, water, nutrients, proteins, and oxygen [16]. Any change to this sensitive interface, irrespective of its cause (e.g. age, disease,
environmental factors), will affect the precise metabolic
balance [11, 16, 17].
The three main risk factors for AMD are age, environmental risk factors, and genetic predisposition. Age itself
influences the viability of both the RPE cells and Bruch’s membrane [11]. Age has several negative effects on many
intracellular structures of the RPE cells, finally leading to
changes in RPE metabolism [11]. Similarly, Bruch’s membrane suffers from age-related changes, such as thickening
and other structural changes that change its permeability.
Altogether, these age-related changes have the potential
to negatively influence the integrity of the RPE/Bruch’s
membrane interface leading to the accumulation of debris
that ultimately leads to the formation of drusen [11]. However, this assumption still has to be confirmed. According
to Anderson et al. local inflammation as a response to debris accumulation plays a critical role in the formation of
drusen [18]. Analyses of the composition of drusen have
shown, that they are composed of lipids, polysaccharides,
glycosaminoglycans, and proteins [18-20]. Additionally,
many proteins showed oxidative modifications, supporting
the hypothesis that oxidative stress is a further contributor
to the pathophysiology of AMD [20].
The most important environmental risk factors are smoking and diet. Smoking increases the risk to develop AMD
by two- to three-fold. Moreover, there is evidence that
there is a dose-dependent association as well as a reversibility in the case of quitting smoking [21]. Regarding an
individual’s diet, healthy forms, e.g. the Mediterranean
diet, is associated with reduced risk due to the high content of antioxidants and vitamins [22, 23]. In contrast,
high-fat or high glucose/fructose diets represent a significant risk factor for AMD. Both direct influences of the nutritional components, as well as more indirect influences
like dysbiosis of the gut microbiota, are thought to be
associated with AMD formation. The latter is thought to
result in systemic low-grade inflammation [24].
To date, the largest study of the genome-wide association
of AMD revealed 52 gene variants across 34 loci [25].
45 out of 52 were classified as common variants and the
remaining 7 as rare variants. Furthermore, the analyses
showed that the genetic risk is shared between the neovascular and the non-neovascular form of AMD except for
one genetic variant that seems to be exclusive for neovascular AMD [25]. Further enrichment analyses narrowed
down the following molecular mechanisms that could be
affected by the identified gene variants: lipid metabolism,
extracellular matrix organization, and assembly as well
as the complement pathway [25]. The possible role of the
complement system in the pathophysiology of AMD was
recently reviewed by Armento et al. [11]. In summary, increasing evidence supports the involvement of the activation of the alternative pathway of the complement system,
both in a local fashion as well as on a systemic basis.
It is hypothesized that the crucial anatomic site where
AMD pathophysiology begins is the complex of RPE
cells, Bruch’s membrane, and the choroid. In the healthy
retina, this complex does not only mediate the precisely
regulated exchange of nutrients and metabolic waste products, but it also inhibits the activation of the alternative
pathway of the complement system. As soon as the AMD
pathophysiology has been triggered through one or more
of its risk factors, the normal function of the complex is
unbalanced. Consequently, both the integrity of RPE cells
and Bruch’s membrane become more and more impaired.
This leads to a cascade of events that disturb retinal homeostasis: accumulation of metabolic end products, oxidative stress, and activation of the complement system
thereby inducing local inflammation and cell senescence
[11, 26, 27]. The exact temporal relationships between the
degeneration of RPE and photoreceptor cells and changes
in Bruch’s membrane and the choriocapillaris are not yet
clear. The first changes may occur in Bruch’s membrane
or the choriocapillaris, leading to RPE and photoreceptor
degeneration. Alternatively, changes in the RPE and photoreceptors could drive the changes in Bruch’s membrane
and choriocapillaris.
Metformin
Metformin is a synthetic derivative of the naturally occurring galegine from the plant Galega officinalis [1]. Chemically, metformin is a biguanide consisting of two coupled guanidine molecules with some additional substitutes. As a derivative of a naturally occurring molecule, metformin has not been designed to target specific pathways, nor did it go through the regulatory process of preclinical and clinical trials which are mandatory today. After its safety and efficacy had been established, metformin has been used as a glucose-lowering agent since the 1950s [1]. FDA approval followed in 1994 and since the UK Prospective Diabetes Study in 1997 (UKPDS) had demonstrated the beneficial effects of metformin, it has been recommended as first-line treatment for type 2 diabetic patients [2].
Metformin mechanisms of action
The classical antihyperglycemic function of metformin
takes place at multiple sites of action in the body and
through multiple molecular mechanisms that have been
described in detail elsewhere [1, 2]. Briefly, its blood
glucose-lowering ability is a combination of effects that
metformin exerts in the liver, the gastrointestinal tract, and
the muscles.
In the liver, gluconeogenesis is downregulated through
both AMPK-dependent and -independent signaling pathways. The AMPK pathway is the cellular energy sensor
and regulator of the cell’s energy homeostasis. If the ratio
of AMP: to ATP increases, the AMPK pathway induces a
switch from ATP-consuming pathways to ATP-generating
pathways. This includes the downregulation of gluconeogenesis and hence, a reduction of glucose levels [1, 2].
In the gastrointestinal tract, metformin is thought to increase glucose uptake and metabolism by colonic enterocytes [1]. Moreover, increased glucagon-like peptide-1
receptor (GLP-1 receptor) secretion has been reported in
response to metformin. Activation of the GLP-1 receptor
results in increased insulin release [2]. Finally, metformin
seems to be related to shifts in the composition of the gut
microbiome, but it remains unclear if and how changes in
the gut microbiome lead to glucose-lowering effects [2].
It is postulated that a healthier gut microbiome suppresses
postprandial hyperglycemia and that levels of inflammatory cytokines are reduced [1].
In skeletal muscles, metformin has been reported to
increase insulin-stimulated uptake of glucose. Newer
investigations, however, indicate that this effect is more
secondary by the metformin-induced overall improvement
of glycemic control and reversal of glucose toxicity [2].
Metformin and AMD
As described above, the pathophysiology of AMD primarily affects the interface of photoreceptors, RPE cells, the choroid, and choriocapillaris. The association of metformin with AMD has been investigated in some preclinical trials, some retrospective trials, and some systematic reviews and meta-analyses based on the reported mechanisms of action of metformin. Before reporting the results of these trials, we will summarize the proposed mechanisms of action of metformin, that could play a role in its influence on the AMD pathophysiology.
Proposed mechanisms of action
The exact mechanisms of the multiple effects of metformin are still under investigation. However, some possible
signaling pathways and/or modes of action have already
been identified.
The AMPK pathway appears to play a central role in the
action of metformin (Figure 2). The AMPK pathway is a
central regulator of cellular metabolism [28]. AMPK becomes activated when the level of ATP decreases indicating high metabolic activity. Via direct phosphorylation of
several proteins, AMPK downregulates energy-consuming
pathways and promotes the activation of energy-producing
pathways to restore the energy homeostasis of the cell [28].
In this way, the AMPK pathway plays a major role in the
regulation of glucose metabolism, lipid metabolism, cell
growth, and autophagy. As described earlier, AMD pathophysiology relies on the integrity of the RPE cells, which
are the critical interface between photoreceptor cells and
the choroid. Dysregulation of RPE metabolic pathways,
especially of the AMPK/SIRT1/PGC-1 and of the mTOR
pathway is strongly associated with AMD pathophysiolog
[29]. Metformin directly influences the mitochondrial
respiratory chain thereby inducing the AMP-mediated activation of AMPK, the initial step of the AMPK pathway
[2, 7]. Downstream signaling within the AMPK pathway
is complex. This could explain why the beneficial functions of metformin are as diverse as anti-inflammatory,
anti-oxidative, anti-angiogenic, and anti-apoptotic [30].
The second mode of action is the ability of metformin to
reduce chronic inflammation by improving the metabolic
state. Additionally, several direct anti-inflammatory effects
have been described, although not directly in the context
of AMD but as a general effect of metformin [31]. This
includes decreasing reactive oxygen species and lowering
levels of inflammatory cytokines [31]. Interestingly, in
the context of the acute respiratory distressed syndrome
(ARDS), a common inflammatory condition in severe
COVID-19, metformin has been shown to inhibit the activation of the NLRP3 inflammasome thereby ameliorating
the course of this life-threatening complication [32]. This
is in line with the finding that fluoxetine, a direct inhibitor
of NLRP3, is associated with a reduced risk to develop
AMD. Possibly, metformin is likewise able to prevent
NLRP3 inflammasome activation in RPE cells to prevent
their degeneration [33].
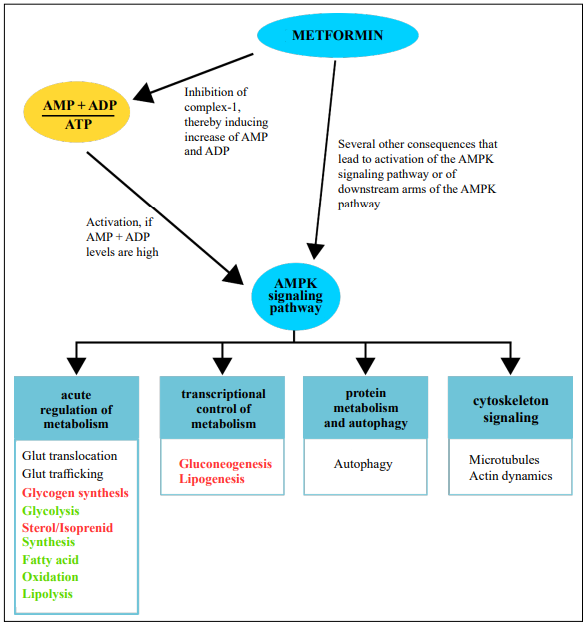
Figure 2. Influence of metformin on the AMPK signaling pathway and consequences of AMPK activation. Without metformin, the AMPK signaling pathway is activated when the cellular levels of AMP and ADP increase. Activation of the pathway leads to a switch from energy-consuming metabolism to energy-providing metabolism. Metformin has been shown to exert parts of its function through the activation of the AMPK pathway. A confirmed mechanism is that metformin can inhibit complex1 of the respiratory chain, thereby inducing the accumulation of AMP and ADP. Furthermore, several other, more direct influences of metformin on downstream components of the AMPK pathway have been reported. AMP = adenosine-monophosphate, ATP = adenosine-triphosphate, ADP = adenosine-diphosphate, AMPK = adenosine-monophosphate dependent kinase, Glut = glucose transporter. Bold red font: inhibition, bold green font: promotion.
Effects of metformin in preclinical trials
The group of Ying et al. investigated the effects of metformin in a mouse model of laser-induced CNV as well
as in the human umbilical vein endothelial cell (HUVEC)
line [34]. Mice treated with metformin had significantly
smaller CNV lesions with reduced vascular density than
the control group. Their experiments with HUVEC cells
showed that activin receptor-like kinase 1 (ALK1), a receptor that is essential for vascular development, remodeling, and pathological angiogenesis, is inhibited by AMPactivated protein kinase (AMPK) and that metformin is a
potent activator of AMPK [34].
The group of Han et al. elucidated the anti-angiogenic and
anti-inflammatory effects of metformin in a set of in-vitro
and in-vivo experiments [35]. They found that metformin
had significant anti-angiogenic effects by inhibiting proliferation, migration, and tube formation of human retinal
vascular endothelial cells. In addition, metformin had potent anti-inflammatory effects by suppressing several inflammatory cytokines through both AMPK-dependent and
AMPK-independent pathways [35]. The authors did not
specify which AMPK-independent pathways are involved
in the mode of action of metformin. However, their experiments showed that suppression of NFB and interleukin-8
by metformin was independent of the AMPK pathway.
Qu et al. examined the effect of metformin on the human
retinal pigment epithelium cell line ARPE-19. Cells were
put under oxidative stress via glyoxal-induced cytotoxicity [36]. Metformin was able to protect ARPE-19 cells by
inhibiting cell death, reducing intracellular reactive oxygen species (ROS) production, decreasing the apoptosis
rate, and increasing intracellular nitric oxide (NO) levels,
an important molecule for maintaining retinal homeostasis
[36]. A subset of experiments confirmed that metformin
influences antioxidant and autophagy pathways to exert its
function [36]. Similar experiments have been performed
by Zhao et al. using two different human pigment epithelium cell lines [37]. Their experiments showed that H2O2-
induced oxidative damage was attenuated by metformin.
Metformin stimulated autophagy via the AMPK pathway
[37].
The in-vivo experiments performed by Xu et al. using different mouse models for retinal and photoreceptor degeneration corroborate the results of the above-described in-vitro experiments [38]. Xu et al. used the albino BALB/
cJ mouse strain to analyze whether metformin can protect
against light-induced photoreceptor loss. If mice were
pretreated with metformin at least 4 days before light
damage was induced via 4h exposure to 4.000 lx bright
white fluorescent light, photoreceptor loss was prevented.
In a subset of experiments, the group used knockout mice
for the AMPK1- and AMPK2-subunit, and showed, that
presence of the 2-subunit was crucial for the protective effect of metformin. As the protection by metformin was the
same between systemic and local (intravitreal) injections,
the authors followed that metformin’s protection is based
on local influences. Xu et al. used a second mouse model,
the Rd10 model for inherited retinal degeneration to analyze the protective effect of metformin. Starting on postnatal day 16, Rd10 mice aggressively lose their rod photoreceptors followed by cone photoreceptor loss. Treatment
with metformin starting on postnatal day 13 delayed the
loss of both photoreceptor types. Via mitochondrial protein expression experiments, Xu et al. could associate
metformin’s protection with increased metabolic activity.
In the third set of experiments, the group injected sodiumiodate into BALB/cJ mice to induce acute oxidative stress
to the RPE and the photoreceptors. This oxidative stress
mimics the early oxidative stress factor of early AMD. If
mice were pretreated with metformin, either 30 or 35 mg/
kg the RPE and photoreceptors were resistant to the damage in a dose-dependent manner: ~50% and ~90% of cells
were protected [38]. None of these mouse models is a perfect model for AMD and such a model does not exist. But
each experiment gives insights into relevant aspects of
the AMD pathophysiology and potential mechanisms that
could be impacted by metformin.
Effects of metformin use on AMD in retrospective clinical trials
Eight retrospective studies have analyzed the association
of metformin use with AMD, of which one was a crosssectional study [39], four were cohort studies [40-43], two
were case-control studies [44, 45], and one was a nested
case-control study [46]. Five studies exclusively determined the association between metformin use and the risk
of developing AMD in diabetic patients [39-43], whereas
three studies included broader patient groups according to
their cohort definition [44-46]. One study examined the
association of metformin use with dry AMD only [43],
while the remaining studies considered all forms of AMD
or did not further specify. All studies took into account
possible confounders like age, sex, ethnicity, smoking
status, insurance status, other oral (antidiabetic) medications, insulin use, cardiovascular disease, hypertension,
hyperlipidemia, obesity, BMI, HbA1c, kidney disease, or
Charlson comorbidity index as far as data were available.
Stewart et al. performed a cross-sectional study using
the electronic medical record database of the University
of California, San Francisco [39]. They included 3,120
diabetic patients who had documented ophthalmologic examinations and a documented metformin use before or at
their first documented ophthalmologic exam. The outcome
of interest was a diagnosis of either non-neovascular or
neovascular AMD at the first ophthalmologic exam. Using propensity score-weighted logistic regression models,
Stewart et al. found that metformin use was significantly
associated with a reduced odd ratio (OR) to develop AMD
(OR 0.70, 95% confidence interval, p-value 0.003). The
association was even stronger when analyzing non-neovascular AMD alone (OR 0.59, 95% CI, p-value < 0.001).
All other antidiabetic drugs studied showed no association. Limitations of this study are the retrospective nature,
the relatively small sample size, the exclusion of drusen
as an early stage of AMD, because authors found the diagnosis of drusen to be unreliable, and missing information
about the duration of metformin use [39].
Chen et al. investigated the association between metformin use and the risk of AMD in a cohort study with type
2 diabetic patients [40]. They included 68,205 patients
who had a diagnosis of type 2 diabetes mellitus during
the study period. Patients were followed up to identify the
onset of AMD (unspecified, non-exudative, or exudative).
The main independent variable was the use of metformin,
which was true for 66.7% of the identified patients. Adjusted hazard ratios (HRs) were obtained via multivariate
Cox regression analyses. Patients taking metformin had a
significantly lower HR to develop AMD than metformin
non-users (HR 0.54, 95% Ci, p-value < 0.0001). Chen et al. also calculated HRs for the duration and cumulative
dose of metformin and their association with the development of AMD and found that both significantly lowered
AMD risk. Limitations of this study are the retrospective nature, and some missing details in the database like
smoking status, diet, and laboratory values [40].
Another cohort study was performed by Jiang et al. [41].
The group reviewed medical records of the ophthalmology department of the China-Japan Friendship Hospital in
Bejing, China. 324 patients with a diagnosis of diabetes
mellitus type 2 for at least 10 years, were identified and
followed-up over 5 years. Patients were excluded if they
had a diagnosis of AMD before the diagnosis of diabetes.
AMD was graded into early and late-stage AMD. Metformin users and non-users were compared using the X2
test and multivariate logistic regression models were used to
characterize the influence of confounders. AMD occurrence in the metformin group was 15.8% and 45.2% in
the metformin non-users (p < 0.0001), thus patients taking metformin had significantly less risk to develop any AMD. Subgroup analysis revealed, that metformin use
only influenced the development of early AMD and not
late AMD. Further analysis showed that both duration of
metformin use and cumulative metformin dose was associated with significantly lower risks to develop any early
but not late AMD. The retrospective design, the small
sample size, and the missing data on some important confounders are the limitations of this study.
Gokhale et al. performed a further cohort study investigating the influence of metformin on the risk of AMD in
patients with type 2 diabetes [42]. The group identified
173,689 patients with newly diagnosed type 2 diabetes
from the United Kingdom IQVIA Medical Research Data.
Patients were excluded if they had an AMD diagnosis
before diabetes diagnosis and if they had no prescription
for antidiabetic medication. 89% of the identified patients
had a prescription for metformin alone or in combination
with other antidiabetic drugs. The control group had any
medication except metformin. The outcome of interest
was a diagnosis of AMD during the study period. HRs
were defined in a time-dependent manner using extended
Cox proportional hazard models. For the time-dependent
analysis, the follow-up intervals were set to 3 months.
AMD occurred in 3,111 (1.8%) of the patients. Gokhale
et al. did not find an association between metformin and
the development of AMD. This finding was independent
of the use of other antidiabetic drugs as well as from the
duration of diabetes and the duration of metformin use.
Limitations of this study include the retrospective design,
and the missing differentiation between AMD stages (early,
late) that could for example mask findings if metformin
was only protective for certain AMD stages [42].
The group of Eton et al. investigated the association of
metformin and dry AMD only [43]. In their cohort study,
they included patients with a diagnosis of diabetes mellitus and sufficient follow-up visits. Furthermore, Eton et
al. distinguished between the current and historical use of
metformin. Current metformin use was defined as metformin use during the study period, historical metformin
use was based on any metformin use before the patients’enrollment date (defined as aged 55 years or more, a diagnosis of diabetes mellitus and at least two years of followup data). Current metformin use was associated with a
small, but significantly increased HR to develop dry AMD
(HR 1.08; 95% CI, p < 0.0001). Historical metformin use,
however, showed a protective effect (HR 0.95; 95% CI, p
= 0.002). The analysis of the cumulative dose of metformin revealed slightly decreased HRs for cumulative doses
below 720,000 mg, but slightly increased HRs for cumulative doses above 720,000 mg. Overall, the study by Eton
et al. showed conflicting results for the effect of metformin on the development of dry AMD. Study limitations
include the retrospective design, potentially the restriction
to dry AMD only, and a probable observation bias [43].
Lee et al. used a different study design, a nested casecontrol study, and they also had a broader definition for
the study eligibility as they not only included patients
with a diagnosis of diabetes mellitus type 1 and 2 but also
patients with a diagnosis of cardiovascular disease [46].
Above that, they were not only interested in the effect of
metformin, but also the effects of statins, angiotensin-converting enzyme (ACE)-inhibitors, and angiotensin II receptor blockers on AMD. During the study, 2,330 patients
developed AMD. For each case, 10 controls were matched
by sex, age, and cohort entry date, leading to a control
group of 23,278 patients. Study outcomes were, that none
of the investigated drugs had a protective effect on the development of AMD. These findings were independent of
the duration of drug use. The nested case-control design
overcomes some of the disadvantages of traditional casecontrol studies, such as the reduction of selection bias.
The retrospective design and its disadvantages remain,
and sample sizes were relatively small.
Two case-control studies investigated the effect of metformin on AMD independent of a diagnosis of diabetes [44, 45]. However, both studies examined diabetic patients
separately as subgroups of the initial total study cohort.
Cases were defined as patients who had a diagnosis of
AMD during the study period. Brown et al. included
patients with all types of AMD (non-exudative, exudative, or unspecified), controls had no AMD and were
propensity score matched using age, Charlson comorbidity index (CCI), hypertension, and anemia as matching
variables [45]. They found that metformin was associated
with statistically significant decreased odds of developing AMD (OR 0.58; 95% CI, p = 0.0005). Other diabetic
and non-diabetic medications showed no association with
AMD. The subgroup analysis of diabetic patients taking
metformin versus non-metformin users showed that metformin was significantly associated with decreased odds
of developing AMD in univariate and multivariate logistic
regression (OR 0.68; 95% Ci, p = 0.002 and OR 0.7; 95%
CI, p = 0.043). Blitzer et al. defined their study cohort
as patients with newly diagnosed AMD during the study
period and powered their study to detect ORs of 0.95 with
90% power in a subgroup of diabetic patients [44]. Controls were selected 1:1 and matched based on age, anemia,
hypertension, region, and CCI score. The effects of diabetes were tested after control matching. Metformin use was
similar in the case and control groups (12.8% and 13.0%).
Use of any metformin was significantly associated with
decreased odds of developing AMD (OR 0.94; 95% CI,
p < 0.001). In addition, it was found that low to moderate total metformin doses had a dose-dependent effect, while there was no association between high metformin (>
1080 g cumulative dose) doses and AMD. The subgroup
analyses of diabetic patients showed similar results. Metformin use significantly decreased the odds of developing
AMD (OR 0.95; 95% CI, p < 0.001), and again a dosedependent effect for low to medium cumulative metformin
doses was found.
In summary, five out of eight retrospective studies found
associations of metformin with decreased odds of developing AMD [39-41, 44, 45], one study found conflicting
associations [43], and two studies report no association
of metformin use with the development of AMD [42, 46].
Three studies found positive associations with either the
duration of metformin use or dose-dependent effects [40, 41,44], while one study did not detect an association with
longer metformin use [46]. In addition, a meta-analysis
by Romdhoniyyah et al. over five of the above-reported
retrospective trials did not find a significant association
between metformin use and the risk to develop AMD [3].
In contrast, a very recently published meta-analysis by
Mauschitz et al. over 14 European population- or hospitalbased studies found a lower AMD prevalence in patients
taking lipid-lowering and/or antidiabetic drugs (including
metformin) (OR 0.78; 95% CI, p = 0.002) [47]. However,
no association was found for late AMD stages (OR 1.12,
95% CI, p = 0.37).
The main limitation of all retrospective studies is that they
can only detect associations but cannot determine causal
relationships. The latter is only possible in the context
of prospective trials. In addition, retrospective trials are
prone to other limitations such as selection bias, recall
bias, loss to follow-up, and confounding factors [48].
Nevertheless, the majority of the described retrospective
analyses found that metformin was associated with decreased odds to develop AMD. Selection bias is especially
small for cohort studies like those of Chen et al., Jiang et
al., Gokhale et al., and Eton et al. [40-43, 48]. All eight studies considered confounding factors and comorbidities
in their analyses. The limitation of loss to follow-up was
reduced by adjusting the eligibility criteria and only patients for whom sufficient follow-up visits were available
were allowed to enter the study cohorts. As metformin is
predominantly described to type 2 diabetic patients, five
studies exclusively investigated the effect of metformin on
AMD in diabetic patients. Three studies included broader
patient groups. Two of them found that metformin decreased the odds of developing AMD independently from
a diagnosis of diabetes. This suggests that diabetes itself
probably has little influence on the development of AMD.
Prospective clinical trials
There is one ongoing prospective, phase II clinical trial that is investigating the ability of metformin to decrease the progression of geographic atrophy (GA) in nondiabetic patients with AMD [49]. A planned population of 186 subjects will be stratified 1:1 into a treatment and an observation group. The treatment group will be assigned to oral metformin for 18 months. At an additional follow-up visit at month 24, the progression of geographic atrophy will be measured and compared between groups. The primary outcome measures are the change in the area of GA or drusen growth. Secondary outcome measures include best-corrected and low-luminance visual acuity, ocular and systemic safety of metformin use, and score changes of the National Eye Institute Visual Function Questionnaire. Subjects with type 1 or 2 diabetes are excluded from the study as well as subjects that are already taking metformin for other reasons. Study completion is expected by the end of 2024.
Limitations of metformin use
Beyond all the reported beneficial properties of metformin, there are also some disadvantages associated with the
use of metformin that should be taken into account before
using metformin as a “cure it medication”. Reported disadvantages include vitamin B12 deficiency, increased risk
of lactic acidosis, and alteration of 745 proteins with uncertain consequences [7]. In addition, metformin is known
to have various gastrointestinal side effects.
Furthermore, a study by Ebeling et al. that analyzed the
influence of metformin on individual patient-derived
RPE cell lines indicated that the effect of metformin was
not uniform across all patients. The group suggests that
patient-specific responsiveness to metformin should be
taken into account before prescription and that approaches
toward personalized medicine are necessary [50].
Conclusion
Evidence is increasing that metformin, the most commonly prescribed oral antihyperglycemic drug, influences
a variety of physiological functions besides its classical
glucose-lowering effect. Essentially, this includes antiinflammatory, anti-angiogenic, anti-oxidative, anti-apoptotic, neuroprotective, and anti-aging effects.
The ongoing prospective trial about the effect of metformin on the progression of geographic atrophy could deliver the first results for this subgroup of late-stage AMD
patients. In the future, more prospective trials are needed
to confirm in more detail how the beneficial effects of
metformin influence the pathophysiology of AMD and
if metformin qualifies as a treatment option in patients
with a diagnosis of AMD. Additionally, prospective trials
should not only concentrate on late-stage dry AMD but
consider all AMD stages. Jiang et al. [41] found for example, that especially the early stage of AMD was associated
with a beneficial effect of metformin. Finally, prospective
trials should consider patients with and without a diagnosis of diabetes to rule out possible confounding effects of
the diabetic disease.
Declarations
Authors’ contributions
The authors contributed equally to the article.
Conflicts of interest
I declare that I have no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
References
1. Rena G, Hardie DG, & Pearson ER. The mechanisms of action of metformin. Diabetologia, 2017, 60(9): 1577- 1585. [Crossref]
2. LaMoia TE, & Shulman GI. Cellular and Molecular Mechanisms of Metformin Action. Endocr Rev, 2021, 42(1): 77- 96. [Crossref]
3. Romdhoniyyah DF, Harding SP, Cheyne CP, & Beare NAV. Metformin, A Potential Role in Age-Related Macular Degeneration: A Systematic Review and Meta-Analysis. Ophthalmol Ther, 2021, 10(2): 245-260. [Crossref]
4. Campbell JM, Bellman SM, Stephenson MD, & Lisy K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: A systematic review and meta-analysis. Ageing Res Rev, 2017, 40: 31-44. [Crossref]
5. Saisho Y. Metformin and Inflammation: Its Potential Beyond Glucose-lowering Effect. Endocr Metab Immune Disord Drug Targets, 2015, 15(3): 196-205. [Crossref]
6. Amin SV, Khanna S, Parvar SP, Shaw LT, Dao D, Hariprasad SM, et al. Metformin and retinal diseases in preclinical and clinical studies: Insights and review of literature. Exp Biol Med (Maywood), 2022, 247(4): 317-329. [Crossref]
7. Soukas AA, Hao H, & Wu L. Metformin as Anti-Aging Therapy: Is It for Everyone? Trends Endocrinol Metab, 2019, 30(10): 745-755. [Crossref]
8. Wong WL, Su X, Li X, Cheung CM, Klein R, Cheng CY, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health, 2014, 2(2): e106-116. [Crossref]
9. Stahl A. The Diagnosis and Treatment of Age-Related Macular Degeneration. Dtsch Arztebl Int, 2020, 117(29- 30): 513-520. [Crossref]
10. Thomas CJ, Mirza RG, & Gill MK. Age-Related Macular Degeneration. Med Clin North Am, 2021, 105(3): 473- 491. [Crossref]
11. Armento A, Ueffing M, & Clark SJ. The complement system in age-related macular degeneration. Cell Mol Life Sci, 2021, 78(10): 4487-4505. [Crossref]
12. Ferris FL, 3rd, Wilkinson CP, Bird A, Chakravarthy U, Chew E, Csaky K, et al. Clinical classification of age-related macular degeneration. Ophthalmology, 2013, 120(4): 844-851. [Crossref]
13. Schmidt-Erfurth U, Chong V, Loewenstein A, Larsen M, Souied E, Schlingemann R, et al. Guidelines for the management of neovascular age-related macular degeneration by the European Society of Retina Specialists (EURETINA). Br J Ophthalmol, 2014, 98(9): 1144-1167. [Crossref]
14. Augustin AJ, Atorf J. Chirurgie bei Makulablutung: Wann und unter welchen Umständen? Ophthalmo-Chirurgie. 2022, 34(1): 27–31. German.
15. Hurley JB. Retina Metabolism and Metabolism in the Pigmented Epithelium: A Busy Intersection. Annu Rev Vis Sci, 2021, 7: 665-692. [Crossref]
16. Cunha-Vaz J, Bernardes R, & Lobo C. Blood-retinal barrier. Eur J Ophthalmol, 2011, 21 Suppl 6: S3-9. [Crossref]
17. Gu X, Neric NJ, Crabb JS, Crabb JW, Bhattacharya SK, Rayborn ME, et al. Age-related changes in the retinal pigment epithelium (RPE). PLoS One, 2012, 7(6): e38673. [Crossref]
18. Anderson DH, Mullins RF, Hageman GS, & Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol, 2002, 134(3): 411-431. [Crossref]
19. Wang L, Clark ME, Crossman DK, Kojima K, Messinger JD, Mobley JA, et al. Abundant lipid and protein components of drusen. PLoS One, 2010, 5(4): e10329. [Crossref]
20. Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A, 2002, 99(23): 14682-14687. [Crossref]
21. Thornton J, Edwards R, Mitchell P, Harrison RA, Buchan I, & Kelly SP. Smoking and age-related macular degeneration: a review of association. Eye (Lond), 2005, 19(9): 935-944. [Crossref]
22. Raimundo M, Mira F, Cachulo MDL, Barreto P, Ribeiro L, Farinha C, et al. Adherence to a Mediterranean diet, lifestyle and age-related macular degeneration: the Coimbra Eye Study - report 3. Acta Ophthalmol, 2018, 96(8): e926-e932. [Crossref]
23. Merle BMJ, Colijn JM, Cougnard-Grégoire A, de KoningBackus APM, Delyfer MN, Kiefte-de Jong JC, et al. Mediterranean Diet and Incidence of Advanced Age-Related Macular Degeneration: The EYE-RISK Consortium. Ophthalmology, 2019, 126(3): 381-390. [Crossref]
24. Rinninella E, Mele MC, Merendino N, Cintoni M, Anselmi G, Caporossi A, et al. The Role of Diet, Micronutrients and the Gut Microbiota in Age-Related Macular Degeneration: New Perspectives from the Gut⁻Retina Axis. Nutrients, 2018, 10(11). [Crossref]
25. Fritsche LG, Igl W, Bailey JN, Grassmann F, Sengupta S, Bragg-Gresham JL, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet, 2016, 48(2): 134-143. [Crossref]
26. Romero-Vazquez S, Llorens V, Soler-Boronat A, FiguerasRoca M, Adan A, & Molins B. Interlink between Inflammation and Oxidative Stress in Age-Related Macular Degeneration: Role of Complement Factor H. Biomedicines, 2021, 9(7). [Crossref]
27. Nita M, & Grzybowski A. Interplay between reactive oxygen species and autophagy in the course of age-related macular degeneration. Excli j, 2020, 19: 1353-1371. [Crossref]
28. Mihaylova MM, & Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol, 2011, 13(9): 1016-1023. [Crossref]
29. Zhang M, Jiang N, Chu Y, Postnikova O, Varghese R, Horvath A, et al. Dysregulated metabolic pathways in agerelated macular degeneration. Sci Rep, 2020, 10(1): 2464. [Crossref]
30. Dang KR, Wu T, Hui YN, & Du HJ. Newly-found functions of metformin for the prevention and treatment of agerelated macular degeneration. Int J Ophthalmol, 2021, 14(8): 1274-1280. [Crossref]
31. Bharath LP, & Nikolajczyk BS. The intersection of metformin and inflammation. Am J Physiol Cell Physiol, 2021, 320(5): C873-c879. [Crossref]
32. Xian H, Liu Y, Rundberg Nilsson A, Gatchalian R, Crother TR, Tourtellotte WG, et al. Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity, 2021, 54(7): 1463-1477.e1411. [Crossref]
33. Ambati M, Apicella I, Wang SB, Narendran S, Leung H, Pereira F, et al. Identification of fluoxetine as a direct NLRP3 inhibitor to treat atrophic macular degeneration. Proc Natl Acad Sci U S A, 2021, 118(41). [Crossref]
34. Ying Y, Ueta T, Jiang S, Lin H, Wang Y, Vavvas D, et al. Metformin inhibits ALK1-mediated angiogenesis via activation of AMPK. Oncotarget, 2017, 8(20): 32794- 32806. [Crossref]
35. Han J, Li Y, Liu X, Zhou T, Sun H, Edwards P, et al. Metformin suppresses retinal angiogenesis and inflammation in vitro and in vivo. PLoS One, 2018, 13(3): e0193031. [Crossref]
36. Qu S, Zhang C, Liu D, Wu J, Tian H, Lu L, et al. Metformin Protects ARPE-19 Cells from Glyoxal-Induced Oxidative Stress. Oxid Med Cell Longev, 2020, 2020: 1740943. [Crossref]
37. Zhao X, Liu L, Jiang Y, Silva M, Zhen X, & Zheng W. Protective Effect of Metformin against Hydrogen PeroxideInduced Oxidative Damage in Human Retinal Pigment Epithelial (RPE) Cells by Enhancing Autophagy through Activation of AMPK Pathway. Oxid Med Cell Longev, 2020, 2020: 2524174. [Crossref]
38. Xu L, Kong L, Wang J, & Ash JD. Stimulation of AMPK prevents degeneration of photoreceptors and the retinal pigment epithelium. Proc Natl Acad Sci U S A, 2018, 115(41): 10475-10480. [Crossref]
39. Stewart JM, Lamy R, Wu F, & Keenan JD. Relationship between Oral Metformin Use and Age-Related Macular Degeneration. Ophthalmol Retina, 2020, 4(11): 1118- 1119. [Crossref]
40. Chen YY, Shen YC, Lai YJ, Wang CY, Lin KH, Feng SC, et al. Association between Metformin and a Lower Risk of Age-Related Macular Degeneration in Patients with Type 2 Diabetes. J Ophthalmol, 2019, 2019: 1649156. [Crossref]
41. Jiang J, Chen Y, Zhang H, Yuan W, Zhao T, Wang N, et al. Association between metformin use and the risk of agerelated macular degeneration in patients with type 2 diabetes: a retrospective study. BMJ Open, 2022, 12(4): e054420. [Crossref]
42. Gokhale KM, Adderley NJ, Subramanian A, Lee WH, Han D, Coker J, et al. Metformin and risk of age-related macular degeneration in individuals with type 2 diabetes: a retrospective cohort study. Br J Ophthalmol, 2022. [Crossref]
43. Eton EA, Wubben TJ, Besirli CG, Hua P, McGeehan B, & VanderBeek BL. Association of metformin and development of dry age-related macular degeneration in a U.S. insurance claims database. Eur J Ophthalmol, 2022, 32(1): 417-423. [Crossref]
44. Blitzer AL, Ham SA, Colby KA, & Skondra D. Association of Metformin Use With Age-Related Macular Degeneration: A Case-Control Study. JAMA Ophthalmol, 2021, 139(3): 302-309. [Crossref]
45. Brown EE, Ball JD, Chen Z, Khurshid GS, Prosperi M, & Ash JD. The Common Antidiabetic Drug Metformin Reduces Odds of Developing Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci, 2019, 60(5): 1470- 1477. [Crossref]
46. Lee H, Jeon HL, Park SJ, & Shin JY. Effect of Statins, Metformin, Angiotensin-Converting Enzyme Inhibitors, and Angiotensin II Receptor Blockers on Age-Related Macular Degeneration. Yonsei Med J, 2019, 60(7): 679-686. [Crossref]
47. Mauschitz MM, Verzijden T, Schuster AK, Elbaz H, Pfeiffer N, Khawaja A, et al. Association of lipid-lowering drugs and antidiabetic drugs with age-related macular degeneration: a meta-analysis in Europeans. Br J Ophthalmol, 2022. [Crossref]
48. Röhrig B, du Prel JB, Wachtlin D, & Blettner M. Types of study in medical research: part 3 of a series on evaluation of scientific publications. Dtsch Arztebl Int, 2009, 106(15): 262-268. [Crossref]
49. University of California, San Francisco. Metformin for the Minimization of Geographic Atrophy Progression in Patients With AMD (METforMIN). 2022 [cited 2022 Aug 12]. Available from: https://clinicaltrials.gov/ct2/show/NCT02684578?cond=metformin+macular+degeneration&draw=2&rank=1
50. Ebeling MC, Geng Z, Stahl MR, Kapphahn RJ, Roehrich H, Montezuma SR, et al. Testing Mitochondrial-Targeted Drugs in iPSC-RPE from Patients with Age-Related Macular Degeneration. Pharmaceuticals (Basel), 2022, 15(1). [Crossref]