Open Access | Review
This work is licensed under a Creative
Commons Attribution-ShareAlike 4.0 International License.
Vitiligo as a potential degenerative disease: from oxidative stress to cellular senescence
* Corresponding author: Qiang Li
Mailing address: Department of Dermatology, Air Force Medical
Center, PLA, Beijing 100142, China.
Email: 16585260@qq.com
Received: 30 May 2022 / Revised: 22 June 2022 / Accepted: 27 June 2022 / Published: 30 June 2022
DOI: 10.31491/APT.2022.06.083
Abstract
Vitiligo is a depigmentation disorder characterized by the loss of melanocytes in the skin, which is aggravated by oxidative stress. The relationship between oxidative stress and cellular senescence is still unclear despite considerable research on melanocyte senescence in vitiligo in recent years. Many chronic diseases associated with oxidative stress, that is, degenerative diseases, have been shown to ultimately result in cellular senescence due to sustained activation of reactive oxygen species. This study advances research on the pathophysiology of vitiligo and its treatment options by summarizing the role of oxidative stress and melanocyte senescence in vitiligo and investigating the mechanisms behind the interaction of melanocyte senescence with oxidative stress.
Keywords
Vitiligo, melanocytes, oxidative stress, cellular senescence, age
Introduction
Vitiligo is characterized by the death or loss of melanocytes.
Its etiology is complex and unclear [1]. In several
investigations, oxidative stress has been implicated in
the etiology of vitiligo and melanocyte destruction [2].
Oxidative stress causes a redox homeostasis imbalance
in melanocytes, characterized by excessive synthesis and
poor clearance of reactive oxygen species (ROS). Due
to the oxidation-promoting state produced by epidermal
melanocytes and the disruption of internal antioxidant
defenses during melanin synthesis, the melanocytes produce
too much ROS to form hydrogen peroxide (H2O2)
during melanogenesis, leaving the melanocytes vulnerable
to oxidative stress attack [3, 4]. Previous research has
shown that melanocytes in nonlesional skin of vitiligo patients
have abnormal characteristics compared to normal
melanocytes [5-7], including increased susceptibility to
oxidative stress, easy shedding of skin after friction, and increased production of bioactive proteins (e.g., IL-6 and
matrix metalloproteinase-3) of the senescence-associated
secretory phenotype (SASP) [5-7].
Hayflick and Moorhead (1961) were the first to characterize
cellular senescence. Cellular senescence is defined as
the cessation of normal cell division due to cellular stressors
such as DNA damage, pro-inflammatory responses,
mitochondrial malfunction, or telomere shortening [8, 9]. Tissue regeneration, wound healing, and embryonic
development have all been demonstrated to benefit from
senescent cells in vivo [10, 11]. SASP, which comprises
several pro-inflammatory cytokines, chemokines, and
growth factors, is primarily responsible for the negative
impacts of senescent cells [12, 13]. Senescent cells can
also communicate with neighboring cells by transferring
proteins to them. For example, senescent cells can secrete
SASP factors, which can cause paracrine senescence in
normal neighboring cells [13, 14], and long-term exposure
to SASP impairs the regenerative capacity of mouse
keratin-forming cells (KCs) [13]. Senescent cells can also
modulate the immune response and thereby facilitate their
own clearance [15]. Numerous studies have demonstrated
that oxidative stress has an important role in the process
of melanocyte senescence. In this review, we discuss how
ROS are generated, how vitiligo melanocytes respond to
oxidative stress, and the molecular and signaling pathways
by which oxidative stress induces vitiligo melanocyte senescence.
Oxidative and antioxidant systems of cells
Although there is no consensus yet on the exact cause of
vitiligo, oxidative stress is considered one of the most
critical triggers of the disease [2]. Oxidative stress is a
disturbance in redox homeostasis characterized by an
imbalance of pro-oxidants and antioxidants. Oxidative
stress in tissues and cells is always caused by an excess of
ROS, which contain H2O2, hydroxyl radicals, hypochlorous
acid, and H2O2 radicals [16]. Previous studies have
focused on endogenous ROS production due to metabolic
activity, but many environmental stimuli, including cytokines,
ultraviolet (UV) radiation, chemotherapeutic drugs,
high temperatures, and even growth factors, can produce
high levels of ROS that disrupt normal redox homeostasis
and convert cells to a state of oxidative stress [17-19].
On the other hand, ROS can be attributed to a range of
internal stimuli: (a) cellular metabolic processes, which
are inherited, such as melanogenesis, which requires more
energy; and (b) abnormal mitochondrial energy metabolism,
which ultimately leads to cell proliferation, differentiation,
and apoptosis [20, 21]. During melanogenesis, the
production of ROS generates dopaquinone from dopa and
then dopachrome, making melanocytes more susceptible
to oxidative damage [22]. In addition, melanogenesis is
an energy-consuming process that requires large amounts
of adenosine triphosphate (ATP). The biogenesis of ATP
itself is accompanied by the production of ROS in the mitochondria
and the formation of H2O2 in the epidermis [23].
Overall, these changes place the melanocyte at the center
of ROS accumulation.
In addition, a complex system of enzymatic and nonenzymatic
antioxidant defenses, including catalase (CAT),
superoxide dismutase, and glutathione peroxidase, can
counteract and regulate overall ROS levels to maintain
physiological homeostasis [24]. Several studies have also
found that oxidative stress is an important factor of the activation
of DNA damage repair (DDR) and that telomeres
are particularly sensitive to a homeostatic imbalance of
ROS [25, 26]. Mitochondria are a major source of ROS
production and are considered a key component of the
generation and replenishment of DNA damage foci, which
are important effectors of cellular senescence [27, 28].
Thus, under the influence of external and internal factors,
melanocytes can produce more ROS that cause an imbalance
in the intracellular antioxidant system and induce
apoptosis and loss of melanocytes.
Overview of cellular senescence
Cellular senescence occurs in damaged cells and prevents
their proliferation in organisms. Cell damage itself does
not directly lead to obvious signs of aging; but when the
damage accumulates and reaches a certain limit, the cells
stop proliferating, resulting in macroscopic tissue weakness
and a physiologically senescent phenotype [29]. Cellular
senescence is a tumor-suppressive program initiated by many stress signals, including telomere wear, DDR,
oxidative damage, subculture conditions, and abnormal
oncogene activation [30]. The hallmark of cellular senescence
is the permanent inhibition of proliferation, which
cannot be overcome by physiological mitogenic stimuli.
Depending on the method of induction and the mechanisms
involved, cellular senescence can be classified into
three types. The earliest and most intensively studied type
of cellular senescence is replicative senescence (RS),
which is characterized early on by telomere wear and activation
of the p53/p21 pathway [31, 32]. The second type
of cellular senescence is stress-induced premature senescence
(SIPS), which is induced by various stresses, such
as oxidative stress and UV light [33-35]. The third type
is oncogene-induced senescence (OIS), which is overactivated
by several oncogenes, particularly, the RAS gene
[36-38]. SIPS and OIS have no apparent telomere wear,
but they play an important role in the activation of the
p16/p38 pathway [39, 40].
Skin cellular senescence is also accompanied by some
phenotypic changes, such as SASP [41]. Coppe et al.
(2008) first proposed the concept of SASP [42]. They
found that senescent cells can promote carcinogenesis of
adjacent precancerous cells by secreting inflammatory and
oncogene-related factors that they called the SASP. Senescent
cells accumulate in various organs accompanied by
a series of complex SASPs, in which the expression and
secretion of different types of cytokines are significantly
increased. SASPs are the most important environmental
effect of senescent cells. The deleterious effects of senescent
cells are mainly attributed to SASPs, which include
proinflammatory cytokines (e.g., IL-1α, IL-1β, IL-6, and
IL-8), growth factors (e.g., HGF, TGF-β, and GM-CSF),
chemokines (e.g., CXCL-1/3 and CXCL-10), and matrix
remodeling enzymes (e.g., metalloproteinases) [41, 43].
The SASP of senescent human hepatocytes expresses other
unique secretory phenotypes and promotes macrophage
migration in addition to the characteristic factors IL-8 and
IL-6 [44].
Senescent cells can induce paracrine senescence in normal
neighboring cells by secreting the SASP factor [14].
Chronic exposure to SASPs can impair the regenerative
capacity of mouse KCs [13]. Several essential phenotypes
have been used to identify senescent cells. They include (1)
cell cycle arrest in the G1 phase, which is often detected
as a lack of DNA replication; (2) a flattened and enlarged
cell morphology; (3) abnormal activation of lysosomes, as
evidenced by positive staining for senescence-associated
β-galactosidase activity (SA-β-gal) [45-48]; (4) significant
chromatin heterogeneity (senescence-associated heterochromatin
foci, SAHF) [49, 50]; (5) telomere shortening,
but as mentioned above, is not a reliable signal for SIPS
and OIS [34, 51]; and (6) high expression of several cell
cycle repressor genes, such as p16, p53, and p21 [31, 52-54].
Melanocyte senescence
Melanocytes: the main senescent population in the human epidermis
Melanocytes are the main senescent cells in the skin senescence
process. As we age, senescent cells accumulate
in human skin [46, 55]. Most studies on skin cell senescence
have focused on skin fibroblasts, and little is known
about the effect of melanocytes on skin senescence. Recent
reports have shown that melanocytes can express the
senescence marker p16ink4a and accumulate in human
skin [56]. Senescent melanocytes also exhibit other senescence
markers, such as reduced HMGB1 and telomere
dysfunction, but not telomere shortening. Waaijer et al.
found that p16ink4a markers are localized almost exclusively
in melanocytes in the epidermis [56, 57]. Therefore,
melanocytes are the major senescent cell population in the
human epidermis.
RS is not a major factor in melanocyte senescence. Victorelli
et al [58]. found that telomeres in skin senescent
melanocytes were not significantly shortened and that
telomeres were unlikely to occur due to extensive loss
of telomeric repeat sequences. Therefore, the telomere
length was not a limiting factor in melanocyte senescence.
Furthermore, fully differentiated melanocytes in the skin
have an extremely low proliferative capacity in vivo and
are thus unlikely to experience sufficient telomere wear to
induce RS [59]. Oxidative stress disrupts the binding of
certain telomeric protein complexes to telomeres, thereby
providing a novel mechanism for telomere shortening [60].
However, the mechanisms of melanocyte senescence and
whether melanocytes have a causal effect on the phenotype
of skin aging remain unclear.
Senescent melanocytes: inducing senescence in neighboring cells
In the vitiligo skin microenvironment, senescent melanocytes
can induce senescence in neighboring keratinocytes.
Nelson et al. demonstrated that senescent cells can induce
DNA damage and senescence in neighboring healthy cells
through the mechanism of SASP secretion and increased
ROS [61, 62]. Victorelli et al. showed that senescent
melanocytes surrounding KCs exhibited significant telomere
damage, providing evidence of the bystander effect
of senescent melanocytes in human skin [58]. They also
found that the conditioned medium from senescent melanocytes
induced the tumor angiogenesis factor(TAF) in
dermal fibroblasts in vitro, suggesting that the induction
of paracrine TAF is mediated by soluble factors released
from senescent melanocytes. Senescent melanocytes have
also been shown to induce telomere dysfunction in periepidermal
KCs. Dysfunctional telomeres provide a source
of persistent DDR for the keratinocyte population, further
limiting the replicative capacity of the cells [63-65]. However,
the molecular mechanisms and signaling pathways
of senescence inducement by senescent melanocytes in
neighboring healthy cells are not yet fully understood.
Victorelli et al. also showed that the release of IP-10
from senescent melanocytes activated the CXCR3 signaling
pathway in peripheral cells, which would increase
mitochondrial ROS production and lead to telomere dys function [58]. Previous studies have also demonstrated
increased ROS production from stimulation of CXCR3
receptors [66] and that these components of SASP, particularly
TGF-β1, induce paracrine telomere dysfunction in
a ROS-dependent manner [62]. Although the mechanisms
that lead to enhanced mitochondrial ROS production
downstream of the CXCR3 signaling pathway have not
yet been fully elucidated, several studies have demonstrated
that Akt is phosphorylated as a consequence of CXCR3
activation [67, 68]. Indeed, Akt is involved in the signaling
cascade that enhances mitochondrial ROS production
during aging [28].
Interference between senescent skin cells and immune cells
The skin contains many types of immune cells, including
mononuclear phagocytes (MNPs) such as Langerhans
cells (LCs), dendritic cells, macrophages, monocytes, and
T cells [69] .The exact relationship between skin immune
cells and skin stromal cells is not yet correctly understood,
but more evidence suggests that the cellular crosstalk between
aging skin stromal cells and immune cells leads to
the aging phenotype of the skin [70].
Among MNPs, LCs are epidermal dendritic cells with
self-renewal properties. Due to the low expression level
of IL-1 in aged skin, the number of LCs is reduced and
shows reduced cell migration to regional lymph nodes
[71]. Reduced migration of LCs may lead to the activation
of antigen-specific T cells and regulatory T cells and maintain
the skin’s immune homeostasis [72]. Macrophages
and monocytes are two other major classes of MNPs in
the skin. Senescent fibroblasts produce several SASPs,
including the C-C-triggered chemokine ligand 2, which
then recruit prostaglandin E2 to produce monocytes and
inhibit T cell immune responses [70]. Under inflammatory
conditions, skin-infiltrating monocytes are guided to
differentiate into macrophages by a cytokine environment
containing monocyte colony-stimulating factor. These
macrophages release high levels of MMPs and ROS to
reduce the skin’s ECM and contribute to chronic inflammation
[70]. These results strongly suggest that the MNP
of the skin actively promotes inflammation and promotes
the skin aging phenotype. In addition, T cells resident in
the skin express a memory phenotype, called skin-resident
memory T cells [73]. It has been demonstrated that aged
skin cells can increase the ratio of CD4+ to CD8+ T cells
[74]. Further studies are needed to understand the role of
these in inflammation.
Relationship between oxidative stress and melanocyte senescence in vitiligo
Oxidative stress: inducing premature senescence of vitiligo melanocytes
Vitiligo melanocyte senescence is closely related to the
inducement of oxidative stress. Harman et al. (1998) proposed
the senescence radical theory, which suggests that senescence is caused by harmful tissue damage from oxidative
stress-induced ROS, a key factor of the inducement
of melanocyte senescence in vitiligo. There is evidence of
elevated levels of H2O2 in the epidermal environment of
melanocytes and KCs of damaged and undamaged skin
in vitiligo patients [75], which suggests the importance of
oxidative stress in the pathogenesis of vitiligo. High levels
of ROS have been associated with various aspects of melanocyte
damage, including the destruction of their DNA,
lipids, proteins, and structural and functional metabolites
[76]. In addition, ROS-induced oxidative stress causes
widespread abnormal organelle function, disrupts metabolic
pathways, and impairs antioxidant defense mechanisms.
Previous studies have shown that melanocytes may not
be completely absent in damaged skin and that they can
proliferate and be passed on to vitiligo patients in both
lesions and nonlesions. However, disrupted growth of
these melanocytes was observed in an in vitro setting, and
the addition of CAT to the culture medium significantly
improved this situation. Therefore, some researchers have
suggested that increasing H2O2 in lesions may not be sufficient
to kill melanocytes in the early stages of vitiligo but
that H2O2 may eventually destroy these melanocytes in
the late stages of vitiligo [77]. In addition, many chronic
diseases associated with oxidative stress are known as
degenerative diseases, and vitiligo may have similar features.
High doses of H2O2 affect the cellular mitochondrial
function and trigger apoptosis. Low doses of H2O2 induce
cellular senescence and expression of cell cycle proteins
[78, 79]. Thus, in the early stages of vitiligo, impaired melanocyte growth induced by oxidative stress may be due to
premature cellular senescence [33], Furthermore, several
molecular and cellular signaling pathways are involved in
oxidative SIPS-like phenotypes of melanocytes, and skin
biopsies from vitiligo patients have shown a senescent
phenotype, which supports the concept that early vitiligo
may be a degenerative disease [5, 33]. However, the signaling
cascade associated with H2O2-induced premature
senescence of melanocytes is not yet fully understood.
Signaling mechanisms involved in vitiligo melanocyte senescence due to oxidative stress
There is evidence that oxidative stress causes genomic DNA damage and ROS leads to cellular senescence. Many signaling proteins are involved in cellular senescence, such as p53, the mitogen of the p38 protein kinase (MAPK), the nuclear factor kappa-B, the mammalian target of rapamycin (mTOR), the transforming growth factor (TGF) beta (-β), and other signaling channels [80].
(1) p38 signaling pathway
The p38 signaling pathway consists of MAPK and the TGF-activated protein kinase (AKT)-binding protein 1, which inhibits telomerase activity and induces human T cell senescence, proliferation, and expression of T cell receptor (TCR) signaling molecules [81]. p38 MAPK signaling activation induces mTOR-mediated autophagy in senescent CD8+ T cells and enhances telomerase activity [82]. p38 MAPK activation would also trigger a DDRindependent SASP senescence phenotype [83]. Cell proliferation can be enhanced by blocking the p38 and PD1 signaling pathways [82]. Similarly, P38 MAPK inhibitors inhibit the aging of corneal endothelial cells, providing evidence for the treatment of corneal endothelial dysfunction [84]. Hou et al. showed that oxidative stress increases ROS in melanocytes, which activates the ERK1/2 and p38MAPK pathways, and increases the p53-independent cell cycle protein-dependent kinase (CDK) inhibitor p21 (CDKN1A), prompting the blocking of the cell cycle in the M phase and preventing its entry in the G1 phase, which makes the cells incapable of replicating properly and thus, inducing premature melanocyte failure (Figure 1) [85].
(2) p53/p21 and p16
p53/p21 and p16(CDKN2A) signaling are the main pathways that induce melanocyte senescence [33, 81, 86-89]. In response to oxidative stress, p53 may be activated, which in turn activates p21 for senescence induction. However, p21 can also be induced in a non-p53-dependent manner [90, 91]. Previous studies have shown that p16 plays a major role in vitiligo melanocyte senescence [5] or in normal melanocyte senescence [92]. p53 signaling in cellular senescence has been studied for many years, and some proteins promote p53-mediated cellular senescence, such as Aurora B kinase [93], secretory phospholipase A(2) [94], and IFN-γ [95], while other proteins inhibit p53-mediated cellular senescence, such as Sirt2 [96], Hsp27 [97], and MAD2 [98]. p53 is a molecular switch that regulates IGF1-induced premature aging [99]. Shortterm exposure to IGF1 promotes cell proliferation, and long-term exposure induces cellular senescence [100]. In addition, Akt and p21 are required to induce cellular senescence downstream of p53 [100]. The p21 gene is a member of the CLP family of cell cycle-dependent kinase inhibitors located downstream of the p53 gene. Together with p53, p21 can constitute a cell cycle G1 checkpoint that cannot be passed on without repair due to DNA damage, thereby reducing the replication and accumulation of damaged DNA and producing an oncogenic effect [53].
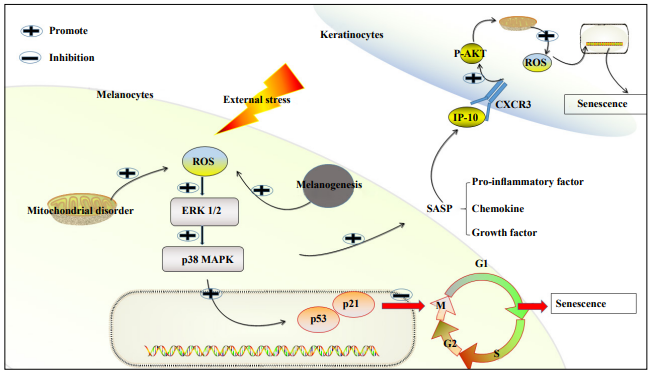
Figure 1. Schematic illustration of oxidative stress-induced skin cell senescence. (1) Oxidative stress increases ROS in melanocytes, which activates the ERK1/2 and p38MAPK pathways and causes the p53-independent cell cycle protein-dependent kinase (CDK) inhibitor p21 (CDKN1A) to increase. This prompts the blocking of the cell cycle in the M phase and prevents its entry in the G1 phase, which makes the cells unable to replicate properly and thus, inducing premature melanocyte failure. (2) The IP-10 released from senescent melanocytes activates the CXCR3 signaling pathway in peripheral keratinocytes, which phosphorylates it by activating Akt and increasing the mitochondrial ROS. This would increase mitochondrial ROS production and lead to telomere dysfunction as well as peripheral keratinocyte senescence.
Antioxidant and antiaging treatment for vitiligo
Vitiligo therapy has always been challenging for dermatologists. The current vitiligo therapy does not appear to be curative. Phototherapy (psoralen mixed with UVA and narrowband UVB [NBUVB]), topical therapies (corticosteroids and calcineurin inhibitors), and systemic treatments (corticosteroids) remain in use, with high side effects to patients and financial burden heavy. Therefore, it is very important to use some anti-oxidant and anti-aging related treatments [4, 101].
Exercise
Several studies have shown that overnutrition significantly increases the expression of senescence-associated proteins, including the activity of p16, p53, p21, and SAβ-gal [102]. Exercise can inhibit the expression of SASPrelated genes and prevent the accumulation of senescent cells caused by overeating [103]. Mechanistically, exercise may reduce the metabolic and replicative stress of adipose tissue and limit the transition to senescence. In addition, exercise may promote the elimination of senescent cells by the immune system [102, 103].
Inhibition of melanocyte senescence by inhibiting oxidative stress
To understand the mechanisms of oxidative stress and cellular senescence in healthy and vitiligo melanocytes and to use these pathways for effective and targeted therapeutic and preventive measures. Natural chemicals with antioxidant potential can inhibit oxidative stress-induced aging. For example, baicalein is a flavonoid derived from the root of Scutellaria baicalensis with anti-cytotoxic, anti-inflammatory, and antitumor effects [104, 105]. In an H2O2-induced oxidative stress model of PIG1 in vitro, baicalein protected PIG1 cells from H2O2-induced oxidative stress and senescence through a mechanism that involved activation of mitochondria-dependent caspases and regulation of the p38MAPK pathway [106]. In H2O2-induced human vitiligo melanocytes (PIG3V), baicalein increased the expression of Nrf2 and its downstream gene HO-1 in the PIG3V cells and promoted the translocation of Nrf2 from the cytoplasm to the nucleus, indicating that the protective effect of baicalein on melanocytes depends on the Nrf2 signaling pathway [107]. Baicalein also has an antioxidant effect on keratinocytes [108]. Therefore, the development of topical formulations of baicalein for vitiligo may be a feasible approach. In addition, some molecules can slow down aging by directly inhibiting ROS production. For example, nicotinamide, an amide derivative of vitamin B3, can slow down aging by reducing the ROS levels [109].
Inhibition of cellular senescence-related pathways
Given that many signaling pathways play an important role in the process of senescence, senescence may be inhibited by inhibiting these pathways. In one study, treatment induced tumor cell senescence, during which Bcl2- associated athanogene 3 (Bag3) increased [110]. Importantly, the knockdown of Bag3 or vault protein (MVP) impairs ERK1/2 activation and promotes treatmentinduced apoptosis in senescent cells [110]. A similar study found that inhibition of the MEK/ERK pathway promotes the clearance of RAS-transformed senescent cells, which prevents these cells from forming the necessary autophagosomes to clear damaged mitochondria and cause apoptosis [111].
Outlook
In summary, we discussed the following aspects. First,
oxidative stress is a key initiating factor of vitiligo. Second, ROS-induced melanocyte senescence in vitiligo is
the major senescent group of skin cells. Third, the IP-10
released from the senescent melanocytes in vivo activates
the CXCR3 signaling pathway in peripheral keratinocytes,
which phosphorylates them by activating Akt, which
increases mitochondrial ROS production, leading to telomere
dysfunction and causing senescence in peripheral keratinocytes.
Fourth, oxidative stress drives increased ROS
in melanocytes, activating the ERK1/2 and p38MAPK
pathways and increasing CDKN1A, which induce premature
melanocyte senescence. Finally, we explored the role
and mechanisms of some antioxidant and antiaging drugs
in vitiligo treatment.
In the context of genetic susceptibility, ROS plays a key
role in the development of vitiligo. ROS contributes to
the destruction of melanocytes in many ways in the early
stages, such as in melanocyte senescence. Furthermore,
cellular senescence plays an important role in both normal
states and physiological conditions. Since its discovery,
many important studies on the role and molecular mechanisms
of cellular senescence have been completed. However,
the role of melanocyte aging in the development
of vitiligo has not been fully elucidated yet. More challenging
questions have been raised. First, the relationship
between cellular senescence and the immune response
remains elusive. For example, it is unclear whether senescent
cells activate adaptive immunity and defend the body.
It is also possible that other novel signaling pathways are
involved in the aging of vitiligo melanocytes. Overall, the
study of cellular senescence is only the beginning, and
there are more interesting questions to be addressed in the
future.
Declarations
Authors’ contributions
Qiang Li is responsible for the direction and overall revision of the article. Yaojun Wang wrote the main content of the manuscript. Jiaoni Chi, Tao Wang, Yue Zhang, Zhimin Li, Jie Chen and Haixia Liu, revised the manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
The authors declare no conflict of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
References
1. Glassman SJ. Vitiligo, reactive oxygen species and T-cells. Clin Sci (Lond), 2011, 120(3): 99-120. [Crossref]
2. Di Dalmazi G, Hirshberg J, Lyle D, Freij JB, & Caturegli P. Reactive oxygen species in organ-specific autoimmunity. Auto Immun Highlights, 2016, 7(1): 11. [Crossref]
3. Denat L, Kadekaro AL, Marrot L, Leachman SA, & AbdelMalek ZA. Melanocytes as instigators and victims of oxidative stress. J Invest Dermatol, 2014, 134(6): 1512- 1518. [Crossref]
4. Chen J, Li S, & Li C. Mechanisms of melanocyte death in vitiligo. Med Res Rev, 2021, 41(2): 1138-1166. [Crossref]
5. Bellei B, Pitisci A, Ottaviani M, Ludovici M, Cota C, Luzi F, et al. Vitiligo: a possible model of degenerative diseases. Plos One, 2013, 8(3): e59782. [Crossref]
6. Gauthier Y, Cario-Andre M, Lepreux S, Pain C, & Taïeb A. Melanocyte detachment after skin friction in non lesional skin of patients with generalized vitiligo. Br J Dermatol, 2003, 148(1): 95-101. [Crossref]
7. Zhou Z, Li CY, Li K, Wang T, Zhang B, & Gao TW. Decreased methionine sulphoxide reductase A expression renders melanocytes more sensitive to oxidative stress: a possible cause for melanocyte loss in vitiligo. Br J Dermatol, 2009,161(3): 504-509. [Crossref]
8. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann NY Acad Sci, 2000, 908: 244-254. [Crossref]
9. Noren Hooten N, Evans MK. Techniques to Induce and Quantify Cellular Senescence. J Vis Exp, 2017, (123): 55533. [Crossref]
10. Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun, 2017, 8: 15691. [Crossref]
11. Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, et al. Programmed cell senescence during mammalian embryonic development. Cell, 2013, 155(5): 1104-1118. [Crossref]
12. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell, 2014, 31(6): 722-733. [Crossref]
13. Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Gene Dev, 2017, 31(2): 172-183. [Crossref]
14. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol, 2013, 15(8): 978-990. [Crossref]
15. Contrepois K, Coudereau C, Benayoun BA, Schuler N, Roux P, Bischof O, et al. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat Commun, 2017, 8: 14995. [Crossref]
16. Wang X, Hai C. Novel insights into redox system and the mechanism of redox regulation. Mol Biol Rep, 2016, 43(7): 607-628. [Crossref]
17. Xie H, Zhou F, Liu L, Zhu G, Li Q, Li C, et al. Vitiligo: How do oxidative stress-induced autoantigens trigger autoimmunity? J Dermatol Sci, 2016, 81(1): 3-9. [Crossref]
18. Hofer S, Stonig M, Wally V, Hartmann A, Fuchs D, Hermann M, et al. Contradictory effects of chemical filters in UV/ROS-stressed human keratinocyte and fibroblast cells. ALTEX, 2019, 36(2): 231-244. [Crossref]
19. Suo D, Zeng S, Zhang J, Meng L, & Weng L. PM2.5 induces apoptosis, oxidative stress injury and melanin metabolic disorder in human melanocytes. Exp Ther Med, 2020, 19(5): 3227-3238. [Crossref]
20. Al-Shobaili HA, Rasheed Z. Oxidized tyrosinase: A possible antigenic stimulus for non-segmental vitiligo autoantibodies. J Dermatol Sci, 2015, 79(3): 203-213. [Crossref]
21. AlGhamdi KM, Kumar A. Depigmentation therapies for normal skin in vitiligo universalis. J Eur Acad Dermatol Venereol, 2011, 25(7): 749-757. [Crossref]
22. Balaban RS, Nemoto S, & Finkel T. Mitochondria, oxidants, and aging. Cell, 2005, 120(4): 483-495. [Crossref]
23. Dell'Anna ML, Ottaviani M, Bellei B, Albanesi V, Cossarizza A, Rossi L, et al. Membrane lipid defects are responsible for the generation of reactive oxygen species in peripheral blood mononuclear cells from vitiligo patients. J Cell Physiol, 2010, 223(1): 187-193. [Crossref]
24. Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol, 1997, 82(2): 291-295. [Crossref]
25. Kruk PA, Rampino NJ, & Bohr VA. DNA damage and repair in telomeres: relation to aging. P Natl Acad Sci USA, 1995, 92(1): 258-262. [Crossref]
26. Rochette PJ, Brash DE. Human telomeres are hypersensitive to UV-induced DNA Damage and refractory to repair. Plos Genet, 2010, 6(4): e1000926. [Crossref]
27. Victorelli S, Passos JF. Telomeres and Cell Senescence - Size Matters Not. Ebiomedicine, 2017, 21: 14-20. [Crossref]
28. Correia-Melo C, Marques FDM, Anderson R, Hewitt G, Hewitt R, Cole J, et al. Mitochondria are required for proageing features of the senescent phenotype. EMBO J, 2016, 35(7): 724-742. [Crossref]
29. Nandi A, Yan L, Jana CK, & Das N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid Med Cell Longev, 2019, 2019: 9613090. [Crossref]
30. Wei W, Ji S. Cellular senescence: Molecular mechanisms and pathogenicity. J Cell Physiol, 2018, 233(12): 9121- 9135. [Crossref]
31. Harley CB, Futcher AB, & Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature, 1990, 345(6274): 458-460. [Crossref]
32. Saretzki G. Telomeres, Telomerase and Ageing. Subcell Biochem, 2018, 90: 221-308. [Crossref]
33. Toussaint O, Medrano EE, & von Zglinicki T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp Gerontol, 2000, 35(8): 927-945. [Crossref]
34. Duan J, Duan J, Zhang Z, & Tong T. Irreversible cellular senescence induced by prolonged exposure to H2O2 involves DNA-damage-and-repair genes and telomere shortening. Int J Biochem Cell Biol, 2005, 37(7): 1407- 1420. [Crossref]
35. CHEN QM. Replicative Senescence and Oxidant-Induced Premature Senescence: Beyond the Control of Cell Cycle Checkpoints. Ann NY Acad Sci, 2000, 908(1): 111-125. [Crossref]
36. Mallette FA, Gaumont-Leclerc M, & Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Gene Dev, 2007, 21(1): 43-48. [Crossref]
37. Scott W, Lowe, Enrique, Cepero, Gerard, & Evan. Intrinsic tumour suppression. Nature, 2004, 432(7015): 307-315. [Crossref]
38. Ferbeyre G, de Stanchina E, Lin AW, Querido E, McCurrach ME, Hannon GJ, et al. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol, 2002, 22(10): 3497-3508. [Crossref]
39. Davis T, Kipling D. Assessing the role of stress signalling via p38 MAP kinase in the premature senescence of ataxia telangiectasia and Werner syndrome fibroblasts. Biogerontology, 2009, 10(3): 253-266. [Crossref]
40. Frippiat C, Dewelle J, Remacle J, & Toussaint O. Free Radic Biol Med, 2002, 33(10): 1334-1346. [Crossref]
41. Coppé J, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol, 2008, 6(12): 2853-2868. [Crossref]
42. Ovadya Y, Krizhanovsky V. Senescent cells: SASPected drivers of age-related pathologies. Biogerontology, 2014, 15(6): 627-642. [Crossref]
43. Bellei B, Picardo M. Premature cell senescence in human skin: Dual face in chronic acquired pigmentary disorders. Ageing Res Rev, 2020, 57: 100981. [Crossref]
44. Irvine KM, Skoien R, Bokil NJ, Melino M, Thomas GP, Loo D, et al. Senescent human hepatocytes express a unique secretory phenotype and promote macrophage migration. World J Gastroentero, 2014, 20(47): 17851-17862. [Crossref]
45. Kurz DJ, Decary S, Hong Y, & Erusalimsky JD. Senescenceassociated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci, 2000, (Pt 20): 3613-3622. [Crossref]
46. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA, 1995, 92(20):9363-9367. [Crossref]
47. Itahana K, Campisi J, & Dimri GP. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol, 2007, 371: 21-31. [Crossref]
48. Shlush LI, Itzkovitz S, Cohen A, Rutenberg A, Berkovitz R, Yehezkel S, et al. Quantitative digital in situ senescenceassociated β-galactosidase assay. BMC Cell Biol, 2011, 12: 16. [Crossref]
49. Kosar M, Bartkova J, Hubackova S, Hodny Z, Lukas J, & Bartek J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a). Cell cycle, 2011, 10(3): 457-468. [Crossref]
50. Zhang R, Chen W, & Adams PD. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol, 2007, 27(6): 2343-2358. [Crossref]
51. von Zglinicki T, Saretzki G, Döcke W, & Lotze C. Mild Hyperoxia Shortens Telomeres and Inhibits Proliferation of Fibroblasts: A Model for Senescence? Exp Cell Res, 1995, 220(1): 186-193. [Crossref]
52. Lewis DA, Yi Q, Travers JB, & Spandau DF. UVB-induced senescence in human keratinocytes requires a functional insulin-like growth factor-1 receptor and p53. Mol Biol Cell, 2008, 19(4): 1346-1353. [Crossref]
53. Shawi M, Autexier C. Telomerase, senescence and ageing. Mech Ageing Dev, 2008, 129(1-2): 3-10. [Crossref]
54. Wang X, Wang Y, & Wang Y. Ginsenoside Rb1, Rg1 and three extracts of traditional Chinese medicine attenuate ultraviolet B-induced G1 growth arrest in HaCaT cells and dermal fibroblasts involve down-regulating the expression of p16, p21 and p53. Photodermatol Photoimmunol Photomed, 2011, 27(4): 203-212. [Crossref]
55. Ressler S, Bartkova J, Niederegger H, Bartek J, Scharffetter-Kochanek K, Jansen-Durr P, et al. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell, 2006, 5(5): 379-389. [Crossref]
56. Waaijer MEC, Gunn DA, Adams PD, Pawlikowski JS, Griffiths CEM, van Heemst D, et al. P16INK4a Positive Cells in Human Skin Are Indicative of Local Elastic Fiber Morphology, Facial Wrinkling, and Perceived Age. J Gerontol A Biol Sci Med Sci, 2016, 71(8): 1022-1028. [Crossref]
57. Waaijer MEC, Parish WE, Strongitharm BH, van Heemst D, Slagboom PE, de Craen AJM, et al. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell, 2012, 11(4): 722-725. [Crossref]
58. Victorelli S, Lagnado A, Halim J, Moore W, Talbot D, Barrett K, et al. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J, 2019, 38(23): e101982. [Crossref]
59. Jimbow K, Roth SI, Fitzpatrick TB, & Szabo G. Mitotic activity in non-neoplastic melanocytes in vivo as determined by histochemical, autoradiographic, and electron microscope studies. J Cell Biol, 1975, 66(3): 663-670. [Crossref]
60. Opresko PL, Fan J, Danzy S, Wilson DMR, & Bohr VA. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res, 2005, 33(4): 1230- 1239. [Crossref]
61. Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell, 2012, 11(2): 345-349. [Crossref]
62. Razdan N, Vasilopoulos T, & Herbig U. Telomere dysfunction promotes transdifferentiation of human fibroblasts into myofibroblasts. Aging Cell, 2018, 17(6): e12838. [Crossref]
63. Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol, 2012, 14(4): 355-365. [Crossref]
64. Fumagalli M, Rossiello F, Mondello C, & d'Adda di Fagagna F. Stable cellular senescence is associated with persistent DDR activation. Plos One, 2014, 9(10): e110969. [Crossref]
65. Hewitt G, Jurk D, Marques FDM, Correia-Melo C, Hardy T, Gackowska A, et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stressinduced senescence. Nat Commun, 2012, 3: 708. [Crossref]
66. Bek MJ, Reinhardt HC, Fischer K, Hirsch JR, Hupfer C, Dayal E, et al. Up-regulation of early growth response gene-1 via the CXCR3 receptor induces reactive oxygen species and inhibits Na+/K+-ATPase activity in an immortalized human proximal tubule cell line. J Immunol, 2003, 170(2): 931-940. [Crossref]
67. Bonacchi A, Romagnani P, Romanelli RG, Efsen E, Annunziato F, Lasagni L, et al. Signal transduction by the chemokine receptor CXCR3: activation of Ras/ERK, Src, and phosphatidylinositol 3-kinase/Akt controls cell migration and proliferation in human vascular pericytes. J Biol Chem, 2001, 276(13): 9945-9954. [Crossref]
68. Shahabuddin S, Ji R, Wang P, Brailoiu E, Dun N, Yang Y, et al. CXCR3 chemokine receptor-induced chemotaxis in human airway epithelial cells: role of p38 MAPK and PI3K signaling pathways. Am J Physiol Cell Physiol, 2006, 291(1): C34-C39. [Crossref]
69. Nestle FO, Di Meglio P, Qin JZ, & Nickoloff BJ. Skin immune sentinels in health and disease. Nat Rev Immunol, 2009, 9(10): 679-691. [Crossref]
70. Lee YI, Choi S, Roh WS, Lee JH, & Kim T. Cellular Senescence and Inflammaging in the Skin Microenvironment. Int J Mol Sci, 2021, 22(8): 3849. [Crossref]
71. Cumberbatch M, Dearman RJ, & Kimber I. Influence of ageing on Langerhans cell migration in mice: identification of a putative deficiency of epidermal interleukin1beta. Immunology, 2002, 105(4): 466-477. [Crossref]
72. Seneschal J, Clark RA, Gehad A, Baecher-Allan CM, & Kupper TS. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity, 2012, 36(5): 873-884. [Crossref]
73. Mueller SN, Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol, 2016, 16(2): 79-89. [Crossref]
74. Zuelgaray E, Boccara D, Ly Ka So S, Boismal F, Mimoun M, Bagot M, et al. Increased expression of PD1 and CD39 on CD3(+) CD4(+) skin T cells in the elderly. Exp Dermatol, 2019, 28(1): 80-82. [Crossref]
75. Schallreuter KU, Salem MA, Holtz S, & Panske A. Basic evidence for epidermal H2O2/ONOO(-)-mediated oxidation/nitration in segmental vitiligo is supported by repigmentation of skin and eyelashes after reduction of epidermal H2O2 with topical NB-UVB-activated pseudocatalase PC-KUS. FASEB J, 2013, 27(8): 3113-3122. [Crossref]
76. Sastry KS,, Naeem H, Mokrab Y, & Chouchane AI. RNAseq Reveals Dysregulation of Novel Melanocyte Genes upon Oxidative Stress: Implications in Vitiligo Pathogenesis. Oxid Med Cell Longev, 2019, 2019: 2841814. [Crossref]
77. Yao L, Hu DN, Chen M, & Li SS. Subtoxic levels hydrogen peroxide-induced expression of interleukin-6 by epidermal melanocytes. Arch Dermatol Res, 2012, 304(10): 831-838. [Crossref]
78. Toussaint O, Dumont P, Remacle J, Dierick J, Pascal T, Frippiat C, et al. Stress-induced premature senescence or stress-induced senescence-like phenotype: one in vivo reality, two possible definitions? Sci World J, 2002, 2: 230-247. [Crossref]
79. lekseenko LL, Zemelko VI, Domnina AP, Lyublinskaya OG, Zenin VV, Pugovkina NA, et al. Sublethal heat shock induces premature senescence rather than apoptosis in human mesenchymal stem cells. Cell Stress Chaperones, 2014, 19(3): 355-366. [Crossref]
80. Martínez-Zamudio RI, Robinson L, Roux P, & Bischof O. SnapShot: Cellular Senescence Pathways. Cell, 2017, 170(4): 816. [Crossref]
81. Lanna A, Henson SM, Escors D, & Akbar AN. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat Immunol, 2014,15(10): 965. [Crossref]
82. Henson SM, Macaulay R, Riddell NE, Nunn CJ, & Akbar AN. Blockade of PD-1 or p38 MAP kinase signaling enhances senescent human CD8(+) T-cell proliferation by distinct pathways. Eur J Immunol, 2015, 45(5): 1441- 1451. [Crossref]
83. Freund A, Patil CK, & Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J, 2011, 30(8): 1536-1548. [Crossref]
84. Akane H, Naoki O, Makiko N, Kay EP, & Noriko K. The Effect of a p38 Mitogen-Activated Protein Kinase Inhibitor on Cellular Senescence of Cultivated Human Corneal Endothelial Cells. Invest Ophthalmol Vis, 2017, 58(9): 3325- 3334. [Crossref]
85. Hou X, Shi J, Sun L, Song L, Zhao W, Xiong X, et al. The involvement of ERK1/2 and p38 MAPK in the premature senescence of melanocytes induced by H(2)O(2) through a p53-independent p21 pathway. J Dermatol Sci, 2022, 105(2): 88-97. [Crossref]
86. Bansal R, Nikiforov MA. Pathways of oncogene-induced senescence in human melanocytic cells. Cell Cycle, 2010, 9(14): 2854-2860. [Crossref]
87. Guterres FADL, Martinez GR, Rocha MEM, & Winnischofer SMB. Simvastatin rises reactive oxygen species levels and induces senescence in human melanoma cells by activation of p53/p21 pathway. Exp Cell Res, 2013, 319(19): 2977-2988. [Crossref]
88. Kuilman T, Michaloglou C, Mooi WJ, & Peeper DS. The essence of senescence. Gene Dev, 2010, 24(22): 2463- 2479. [Crossref]
89. Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J, 2003, 2(16): 4212-4222. [Crossref]
90. Zuo S, Liu C, Wang J, Wang F, Xu W, Cui S, et al. IGFBPrP1 induces p21 expression through a p53-independent pathway, leading to cellular senescence of MCF-7 breast cancer cells. J Cancer Res Clin, 2012, 138(6): 1045-1055. [Crossref]
91. Sayama K, Shirakata Y, Midorikawa K, Hanakawa Y, & Hashimoto K. Possible involvement of p21 but not of p16 or p53 in keratinocyte senescence. J Cell Physiol, 1999, 179(1): 40-44. [Crossref]
92. viderskaya EV, Gray-Schopfer VC, Hill SP, Smit NP, EvansWhipp TJ, Bond J, et al. p16/cyclin-dependent kinase inhibitor 2A deficiency in human melanocyte senescence, apoptosis, and immortalization: possible implications for melanoma progression. J Natl Cancer Inst, 2003, 95(10): 723-732. [Crossref]
93. Kim H, Cho JH, Quan H, & Kim J. Down-regulation of Aurora B kinase induces cellular senescence in human fibroblasts and endothelial cells through a p53-dependent pathway. FEBS Lett, 2011, 585(22): 3569-3576. [Crossref]
94. Kim HJ, Kim KS, Kim SH, Baek S, Kim HY, Lee C, et al. Induction of cellular senescence by secretory phospholipase A2 in human dermal fibroblasts through an ROSmediated p53 pathway. J Gerontol A Biol Sci Med Sci, 2009, 64(3): 351-362. [Crossref]
95. Kim KS, Kang KW, Seu YB, Baek S, & Kim J. Interferongamma induces cellular senescence through p53-dependent DNA damage signaling in human endothelial cells. Mech Ageing Dev, 2009, 130(3): 179-188. [Crossref]
96. Langley E, Pearson M, Faretta M, Bauer U, Frye RA, Minucci S, et al. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J, 2002, 21(10): 2383-2396. [Crossref]
97. O'Callaghan-Sunol C, Gabai VL, & Sherman MY. Hsp27 modulates p53 signaling and suppresses cellular senescence. Cancer Res, 2007, 67(24): 11779-11788. [Crossref]
98. Lentini L, Barra V, Schillaci T, & Di Leonardo A. MAD2 depletion triggers premature cellular senescence in human primary fibroblasts by activating a p53 pathway preventing aneuploid cells propagation. J Cell Physiol, 2012, 227(9): 3324-3332. [Crossref]
99. Tran D, Bergholz J, Zhang H, He H, Wang Y, Zhang Y, et al. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell, 2014, 13(4): 669-678. [Crossref]
100. Pilpel, Noam, Papismadov, Nurit, Krizhanovsky, Valery, et al. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J, 2017, 36(15): 2280-2295. [Crossref]
101. Frisoli ML, Essien K, & Harris JE. Vitiligo: Mechanisms of Pathogenesis and Treatment. Annu Rev Immunol, 2020, 38: 621-648. [Crossref]
102. Schafer MJ, White TA, Evans G, Tonne JM, Verzosa GC, Stout MB, et al. Exercise Prevents Diet-Induced Cellular Senescence in Adipose Tissue. Diabetes, 2016, 65(6): 1606-1615. [Crossref]
103. de França E, Dos SR, Baptista LC, Da SM, Fukushima AR, Hirota VB, et al. Potential Role of Chronic Physical Exercise as a Treatment in the Development of Vitiligo. Front Physiol, 2022,13: 843784. [Crossref]
104. Pang Y, Wu S, He Y, Nian Q, Lei J, Yao Y, et al. Plant-Derived Compounds as Promising Therapeutics for Vitiligo. Front Pharmacol, 2021, 12: 685116. [Crossref]
105. Huang W, Lee A, Chien P, & Chou T. Synthesis of baicalein derivatives as potential anti-aggregatory and antiinflammatory agents. J Pharm Pharmacol, 2005, 57(2): 219-225. [Crossref]
106. Liu B, Jian Z, Li Q, Li K, Wang Z, Liu L, et al. Baicalein protects human melanocytes from H2O2-induced apoptosis via inhibiting mitochondria-dependent caspase activation and the p38 MAPK pathway. Free Radic Biol Med, 2012, 53(2): 183-193. [Crossref]
107. Ma J, Li S, Zhu L, Guo S, Yi X, Cui T, et al. Baicalein protects human vitiligo melanocytes from oxidative stress through activation of NF-E2-related factor2 (Nrf2) signaling pathway. Free Radic Biol Med, 2018, 129: 492-503. [Crossref]
108. Wang Y, Cho J, Hwang E, Yang J, Gao W, Fang M, et al. Enhancement of Protective Effects of Radix Scutellariae on UVB-induced Photo Damage in Human HaCaT Keratinocytes. Appl Biochem Biotech, 2018, 184(4): 1073-1093. [Crossref]
109. Kwak JY, Ham HJ, Kim CM, & Hwang ES. Nicotinamide exerts antioxidative effects on senescent cells. Mol Cells, 2015, 38(3): 229-235. [Crossref]
110. Pasillas MP, Shields S, Reilly R, Strnadel J, Behl C, Park R, et al. Proteomic analysis reveals a role for Bcl2-associated athanogene 3 and major vault protein in resistance to apoptosis in senescent cells by regulating ERK1/2 activation. Mol Cell Proteomics, 2015, 14(1): 1-14. [Crossref]
111. Kochetkova EY, Blinova GI, Bystrova OA, Martynova MG, Pospelov VA, & Pospelova TV. Targeted elimination of senescent Ras-transformed cells by suppression of MEK/ ERK pathway. Aging, 2017, 9(11): 2352-2375. [Crossref]