Open Access | Case Report
This work is licensed under a Creative
Commons Attribution-ShareAlike 4.0 International License.
Nasal type extranodal natural killer/T-cell lymphomaassociated hemophagocytic lymphohistiocytosis: A Case report and literature review
* Correspondence: Liang Wang
Mailing address: Department of Hematology, Beijing Tongren Hospital, Capital Medical University, Beijing 100730, China.
Email: wangliangtrhos@126.com
Received: 11 February 2022 / Accepted: 02 March 2022
DOI: 10.31491/APT.2022.03.077
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a clinical syndrome in which a large number of inflammatory cytokines, such as IL-6, TNF-α, and sCD25, are released into the blood circulation due to cytotoxic killing cells and dysfunction of natural killer (NK) cells. HLH can be induced by various infections, cancer, or hereditary disorders. HLH is usually fatal due to severe cytopenia and multiple organ failure. Nasal type extranodal natural killer/T-cell lymphoma (ENKTCL) is an aggressive non-Hodgkin’s lymphoma that is closely related to Epstein-Barr virus. ENKTCL can be complicated by HLH at initial diagnosis and recurrence. However, the optimal treatment strategies for ENKTCL-associated HLH are not well defined. Here, we report a case of ENKTCL-associated HLH that was successfully treated with liposomal doxorubicin, etoposide, methylprednisolone, and pegaspargase (DEPL regimen), followed by immunotherapy. This report aims to contribute to improved recognition, diagnosis, and treatment of lymphoma-associated HLH.
Keywords
Hemophagocytic lymphohistiocytosis, extranodal natural killer/T-cell lymphoma, liposomal doxorubicin, pegaspargase, lymphoma-associated HLH
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a kind of histiocytosis caused by reactive hyperplasia of the mononuclear macrophage system, mainly due to cytotoxic lymphocytes (CTL, primarily CD8+T cells) and defects in the function of natural killer (NK) cells. These issues then lead to antigen clearance disorder. HLH is a heterogeneous clinical syndrome. During HLH, the monocytemacrophage system receives constant antigen stimulation, resulting in hyperactive proliferation. Then, large inflammatory cytokines are produced, such as IL-6, TNF-α, and sCD25. The primary symptoms include fever, splenomegaly, thrombocytopenia, hypertriglyceridemia, hypofibrinogenemia, and elevated serum ferritin. Hemophagocytes may appear in bone marrow, spleen, or lymph node biopsies [1]. HLH may be primary or secondary. The former is usually caused by gene abnormalities and often occurs in children without underlying diseases. The latter is often secondary to other diseases such as severe infection, advanced malignancies, or serious autoimmune diseases. In a study of 2,197 adults with HLH, the most common primary diseases were T cell or NK cell lymphomas (16.8%), B cell lymphomas (15.2%), Epstein-Barr virus (15.0%), HIV (7.9%), systemic lupus erythematosus (6.1%), and mycobacterium tuberculosis infection (3.6%) [2].
Nasal type extranodal natural killer/T-cell lymphoma (ENKTCL) is an aggressive non-Hodgkin’s lymphoma closely that is associated with Epstein-Barr virus. Approximately 90% of primary lesions occur in the nasal cavity. HLH can be complicated at initial diagnosis, recurrence, and progression; usually responds poorly to treatment; and has a poor prognosis. To contribute to improved recognition, diagnosis, and management of lymphoma-associated HLH, here, we review existing literature on this illness and report one case of ENKTCL-associated HLH, summarizing the case’s clinical characteristics and treatment.
Case report
A 36-year-old woman presented with a one-month history of a mass on her left eyelid and fever (Figure 1A). On April 20, 2021, she developed redness and swelling in the left inferior palpebra along with intermittent fever, with a maximum temperature of 39.5℃. She was admitted to a nearby hospital, where she received topical treatment with antibiotics and glucocorticoids. However, no significant improvement was observed. The mass on the left palpebrae inferior gradually enlarged, leading to impaired vision. Multiple nodules were also found in both breasts. Orbital magnetic resonance imaging (MRI) showed left palpebrae inferior incrassation and lacrimal gland enlargement with an abnormal signal in the right orbit; this was diagnosed as a lymphoproliferative lesion. Positron emission tomography/computed tomography (PET/CT) showed multiple systemic lymphadenopathies and multiorgan space-occupying lesions with a significant amount of metabolic activity, suggesting a high possibility of lymphoma (Figure 2A).
The patient was admitted to our department on May 21, 2021. A physical exam showed an Eastern Cooperative Oncology Group’s performance status (ECOG-PS) score of 2, a temperature of 38.8℃, and multiple enlarged, superficial, palpable lymph nodes. She also had severe redness and swelling in the left inferior palpebrae, where there was a surface ulcer that was about 0.5 cm × 0.3 cm. A 1.5 cm × 1.0 cm mass was palpable. There were purulent secretions in the conjunctiva of the right eye and multiple palpable nodules in both breasts. The largest of these, with a diameter of 2.0 cm, was located in the left outer lower quadrant. This nodule was sensitive to pressure; erythema could be seen on the surface of both breasts as well. The liver was palpable under the costa and was about 8 cm from the costal margin. The spleen was also palpable under the costa and was also 8 cm from the costal margin. A complete blood count (CBC) was conducted; the patient’s white blood cell count was 4.3 × 109/L (normal range: 3.5–9.5 × 109/L), hemoglobin was 98 g/L (normal range: 110–165 g/L), and platelets were 176×109/L (normal range: 125–350 × 109/L). Biochemical tests were also run; her LDH was 504 U/L (normal range: 120–250 U/L). Her hepatic or nephritic function was essentially normal. Her serum ferritin was elevated at 1570 ng/ml (normal range: 15–200 ng/mL). Bone marrow flow cytometry showed that suspected abnormal NK cells accounted for 15.58%; these cells had immunophenotypes of CD3-, CD16-, and CD56+. A bone marrow pathology examination did not reveal any definite tumor cells. NK cell activity was decreased to 14.94%. Her soluble CD25 was over 44,000 pg/ml (normal range:< 6,400 pg/mL). Quantitative detection of EBV-DNA revealed 1.412 × 105 copies/mL (normal range: < 500 copies/mL).
Based on the patient’s continuous fever, hepatosplenomegaly, elevated serum ferritin, decreased NK cell activity, and elevated sCD25, HLH was diagnosed, with probable lymphoma-associated HLH. An enlarged lymph node on the right side of her neck was biopsied, and a DEP regimen (40 mg liposomal doxorubicin on day 1, 150 mg etoposide on day 1, 900 mg methylprednisolone from days 1 to 3, 40 mg methylprednisolone from days 4 to 7, and 15 mg methylprednisolone from days 8 to 10) was administered starting on May 30, 2021. On June 1, 2021, a pathology examination of the right neck lymph node confirmed the diagnosis of nasal type extranodal NK/T cell lymphoma; the immunophenotypes of the lymphoma cells were CD3 (+), CD20 (-), CD43 (+), Ki-67 (index 85%), C-myc (+), CD56 (+), TIA-1 (+), GrB (+), CD30 (-), ALK (-), and CD38 (-); in-situ hybridization of ISH: EBER was positive. The patient received a definitive diagnosis of ENKTCL with stage IVEB and ENKTCLassociated HLH. Her PINK-E score was 4 (high risk), and her NRI score was 5 (very high risk). Thus, a dose of 3,750 U pegaspargase was added to the DEP regimen (i.e., a DEPL regimen was begun) on the fourth day to enhance the anti-lymphoma effectiveness of her treatment. On June 7, 2021, a lumbar puncture was conducted. Although no obvious abnormalities were identified in the cerebrospinal fluid, 50 mg cytarabine, 5 mg dexamethasone, and 10 mg methotrexate were injected intrathecally as a prophylaxis against central nervous system lymphoma.
After chemotherapy, the patient’s body temperature returned to normal; the mass on her eyelid shrank; her limb, lower back, and perineum edema subsided significantly; her breast nodules almost disappeared, her lower limb pain subsided; and her pericardial effusion gradually decreased. This suggested a positive response to the treatment. On June 18, 2021, the second cycle of the DEPL regimen was administered. After this treatment, the patient’s EBVDNA decreased to 3.669 × 10E4 copies/ml; sCD25 was 12,672 pg/ml, and NK cell activity was 14.84%. The lymphoma lesions also decreased in size. However, due to the extensive number of lesions found at diagnosis, the large tumor load, the diagnosis of lymphoma-associated HLH, and the patient’s other high-risk factors, a regimen of 200 mg tislelizumab (PD-1 monoclonal antibody) once every three weeks was begun on July 9, 2021. The third and fourth chemotherapy regimens of GPED (1.5 g gemcitabine on day 1, 3750 U pegaspargase on day 2, 100 mg etoposide from days 2 to 4, and 20 mg dexamethasone from days 1 to 4), were administrated starting on July 13, 2021, and August 9, 2021. We also conducted two more lumbar punctures and intrathecal injections following the same regimen as previously. Routine tests, biochemical tests, and flow cytometry of cerebrospinal fluid showed no obvious abnormalities. After four cycles of chemotherapy, the patient’s ferritin decreased to 997.3 ng/mL, quantitative detection of EBV-DNA decreased to less than 500 copies/mL, sCD25 was 7916 pg/ml, and NK cell activity was 13.17%. The mass on the patient’s eyelid disappeared (Figure 1B). A PET-CT on August 27, 2021, showed significant reductions in the lymphoma lesions compared to May 20, 2021; it also indicated that metabolic activity had decreased or returned to normal (Figure 2B). The patient had therefore reached partial response according to the Lugano 2014 criteria [3]. As of this writing, the patient is still receiving further treatment and is responding well.
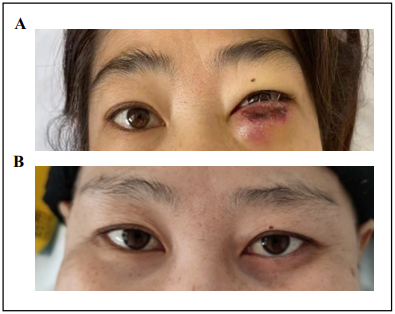
Figure 1. Signs of left palpebra inferior before and after treatment: (A) Severe swelling of the left palpebrae inferior with ulcer and purulent secretions before treatment; (B) Normal left palpebrae inferior after two cycles of treatment.
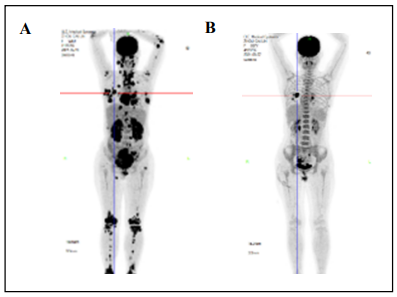
Figure 2. Results of PET-CT before and after treatment: (A) Multiple hypermetabolic lesions were observed in PET/CT before treatment; (B)After two cycles of therapy, PET-CT showed that most of the lesions disappeared or shrank, which metabolic activity returned to normal or decreased, and the treatment response was assessed as partial remission.
Discussion
Malignancy is one of the main primary causes of secondary HLH. The mechanism of malignancy-induced HLH is not fully understood; it is possible that cytokines secreted by malignant tumor cells (including interferon γ and interleukin-6) lead to an excessive inflammatory response. The malignancies that most commonly induce HLH are T cell and NK cell lymphoma or leukemia, diffuse large B cell lymphoma, and Hodgkin’s lymphoma. HLH can occur at any stage of the disease but is more likely in newly diagnosed patients with advanced malignancy and in patients with relapsed or refractory diseases. ElevatedsCD25 is a marker of T cell activation in HLH and an indicator of the tumor load of non-Hodgkin’s lymphoma. In cases of malignancy-induced HLH, viral infection may be a side-by-side trigger, as with EBV-associated lymphoma [4]. When our patient was diagnosed with ENKTCL, extensive lesions were found; she was in clinical stage IV and had significantly elevated EBV-DNA. We therefore concluded that ENKTCL and an EBV infection together induced HLH.
When HLH is secondary to advanced tumors, there is significant overlap between the presentation of HLH and of the tumor, making it very difficult to diagnose HLH. Therefore, an HLH diagnosis is based on a series of clinical manifestations and laboratory tests. According to the HLH-2004 criterion, at least five of the following eight criteria must be met for a diagnosis of HLL [5]: (1) fever: constant fever lasting longer than seven days with a body temperature over 38.5℃; (2) splenomegaly (at least 3 cm subcostal); (3) cytopenia affecting at least two of three lineages in the peripheral blood (hemoglobin < 90 g/L, platelet 100 × 109/L, neutrophil < 1.0 × 109/L) and not caused by bone marrow hematopoietic dysfunction; (4) hypertriglyceridemia and/or hypofibrinogenemia (triglyceridemia > 3 mmol/L or higher than three standard deviations for the patient’s age cohort, fibrinogenemia < 1.5 g/L or lower than three standard deviations for the age cohort); (5) hemophagocytosis in the bone marrow, spleen, or lymph nodes; (6) low or absent NK cell activity; (7) ferritin ≥ 500 μg/L; (8) high levels of sIL-2r (sCD25). Our patient presented with fever, splenomegaly, elevated ferritin, decreased NK cell activity, and elevated sCD25. The diagnosis of HLH was made without controversy. Any patient diagnosed with HLH should be investigated for primary or underlying diseases that might induce HLH [4]. Young patients should also be tested for HLH-related genes to determine whether the HLH is primary. As our patient was diagnosed with ENKTCL and EBV infections, no HLH-related genetic tests were performed.
Secondary HLH is treated by controlling the primary disease as quickly as possible while also actively treating HLH. Primary HLH is treated with allogeneic hematopoietic stem cell transplantation as quickly as possible after HLH has been controlled. The 1994 and 2004 guidelines for HLH induction therapy recommend the use of etoposide combined with dexamethasone. Before treatment, the patient’s primary diseases, physical status, and vital organ functions should be fully examined so that individualized therapy can be provided [4]. Our patient with ENKTCL had a newly diagnosed advanced disease with extensive lesions.
As etoposide combined with dexamethasone performs poorly to simultaneously control HLH and ENKTL, we chose the DEP ± L regimen (liposomal doxorubicin, etoposide, methylprednisolone ± l-asparaginase/pegaspargase). In a multicenter study of the combination DEP regimen to treat adult refractory HLH, etoposide and corticosteroids were used as core agents, the dosage of corticosteroids for pulse therapy was increased during theinitial induction period, and liposomal doxorubicin was used as an important induction therapy. First, glucocorticoids have strong inhibitory effects on immune activation and immunoreaction. Second, doxorubicin, a broad-spectrum cell toxic drug, has strong cell toxicity for a variety of tumor cells. Liposomes can promote drug accumulation in sites where capillary permeability is elevated and can enrich the lymphatic system, prevent the drug from being engulfed by macrophages and monocytes, and prolong the drug’s half-life to ultimately increase the therapeutic effect [6]. Third, etoposide inhibits topoisomerase II, leading to dsDNA breaks; it also suppresses the production of inflammatory cytokine, selectively depletes activated T cells in HLH mice models, and improves survival rates. Therefore, etoposide is a key drug in the HLH-1994 and HLH-2004 guidelines. In cases of lymphoma-associated HLH, both HLH and lymphoma must be treated simultaneously. Several experts suggest that this kind of patient should be treated with etoposide-containing chemotherapy as quickly as possible. Therefore, for our patient with ENKTCL-associated HLH, we adopted the salvage treatment regimen of DEPL for recurrent/refractory HLH, using pegasparagase as the key drug to address ENKTCL [7]. After two cycles of chemotherapy, the patient’s HLH reached partial response. We then quickly switched to treating the primary ENKTCL, using a GPED regimen to address both the ENKTL and HLH, and added a PD-1 monoclonal antibody treatment, which is effective for treating ENKTCL [8-10]. After another two cycles of chemotherapy, a PET-CT showed that the overall tumor load had reduced by more than 80%; there were some residual lesions, mostly in the right breast and ovary. According to the Lugano 2014 criteria, this indicates a partial response to treatment (Figure 2B). No standard treatment has been defined for consolidated treatment of advanced ENKTCLassociated HLH [11]. Previous studies have reported that autologous hematopoietic stem cell transplantation does not significantly reduce the recurrence rate of advanced ENKTCL [12]. If the complete response can be achieved, allogeneic HSCT can be attempted for consolidation therapy in young patients with lymphoma-associated HLH. Our patient is undergoing further treatment, and if the complete response is achieved, allogeneic HSCT will be considered.
HLH has a high mortality rate. For newly diagnosed HLH patients, underlying causes, especially malignancies and infection, must first be confirmed or excluded, and the primary disease must be treated promptly. For lymphomaassociated HLH, a combination chemotherapy regimen containing etoposide and glucocorticoid can be used to treat both HLH and lymphoma at once. However, the overall cure rate of lymphoma-associated HLH is still low, and the prognosis is extremely poor. Therefore, new strategies should be actively explored. Future studies with larger sample sizes should explore the impact of immunochemotherapy regimens that include immune checkpoint inhibitors on the prognosis of ENKTCL patients with HLH.
Declarations
Availability of data and materials
Liang Wang conceived the study; Jin Ye and Jing Yang treated the patient; Xuelin Liu and Liqiang Wei collected the data and wrote the paper. All authors revised the paper and approved the final version.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This research was funded by grants from the National Natural Science Foundation of China (81873450, 82170181) and Beijing Municipal Administration of Hospitals’ Youth Program (code: QMS20200201) to Liang Wang.
Conflicts of interest
Liang Wang is a member of the Editorial Board of Aging Pathobiology and Therapeutics. All authors declare no conflict of interest and were not involved in the journal’s review or desicions related to this manuscript.
Ethical approval and consent to participate
The study, which included human samples, complied with the Declaration of Helsinki and obtained written informed consent from the patient involved.
References
1. Malinowska I, Machaczka M, Popko K, et al. Hemophagocytic syndrome in children and adults. Archivum immunologiae et therapiae experimentalis, 2014, 62(5): 385-394.
2. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al. Adult haemophagocytic syndrome. The Lancet, 2014, 383(9927): 1503-1516.
3. Van Heertum R L, Scarimbolo R, Wolodzko J G, et al. Lugano 2014 criteria for assessing FDG-PET/CT in lymphoma: an operational approach for clinical trials. Drug design, development and therapy, 2017, 11: 1719.
4. Lehmberg K, Nichols K E, Henter J I, et al. Consensus recommendations for the diagnosis and management of hemophagocytic lymphohistiocytosis associated with malignancies. Haematologica, 2015, 100(8): 997.
5. Henter J I, Horne A C, Aricó M, et al. HLH‐2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatric blood & cancer, 2007, 48(2): 124-131.
6. Wang Y, Huang W, Hu L, et al. Multicenter study of combination DEP regimen as a salvage therapy for adult refractory hemophagocytic lymphohistiocytosis. Blood, The Journal of the American Society of Hematology, 2015, 126(19): 2186-2192.
7. Yamaguchi M, Suzuki R, Oguchi M. Advances in the treatment of extranodal NK/T-cell lymphoma, nasal type. Blood, The Journal of the American Society of Hematology, 2018, 131(23): 2528-2540.
8. Tao R, Fan L, Song Y, et al. Sintilimab for relapsed/refractory (r/r) extranodal NK/T-cell lymphoma (ENKTL): A multicenter, single-arm, phase 2 trial (ORIENT-4). 2019.
9. Kwong Y L, Chan T S Y, Tan D, et al. PD1 blockade withpembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood, The Journal of the American Society of Hematology, 2017, 129(17): 2437-2442.
10. Cai J, Liu P, Huang H, et al. Combination of anti-PD-1 antibody with P-GEMOX as a potentially effective immunochemotherapy for advanced natural killer/T cell lymphoma. Signal transduction and targeted therapy, 2020, 5(1): 1-9.
11. Wei L, Yang L, Cong J, et al. Using etoposide+ dexamethasone-based regimens to treat nasal type extranodal natural killer/T-cell lymphoma-associated hemophagocytic lymphohistiocytosis. Journal of Cancer Research and Clinical Oncology, 2021, 147(3): 863-869.
12. Song G Y, Yoon D H, Suh C, et al. Open-label, single arm, multicenter phase II study of VIDL induction chemotherapy followed by upfront autologous stem cell transplantation in patients with advanced stage extranodal NK/T-cell lymphoma. Bone Marrow Transplantation, 2021, 56(5): 1205-1208.