Open Access | Review
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
Aging pet cats develop neuropathology similar to human Alzheimer’s disease
* Corresponding author: Warren Ladiges
Mailing address: Department of Comparative Medicine, School
of Medicine, University of Washington, Seattle, WA, USA.
E-mail: wladiges@uw.edu
Received: 07 September 2020 / Accepted: 11 Septemper 2020
DOI: 10.31491/APT.2020.09.027
Abstract
Aging pet cats can spontaneously develop Aβ deposition and tauopathy (including neurofibrillary tangle formation) with neuronal loss in a similar distribution and with similar characteristics to Alzheimer’s disease (AD) in humans. These three major pathologies that characterize AD rarely occur spontaneously in other nonhuman animals. In addition, cats develop cognitive impairment with increasing age, and some studies show an association with neuronal lesions. These features suggest that the aging pet cat may be a more reliable spontaneously occurring model to investigate pathogenesis of, and therapeutic interventions for, AD compared to other domestic animals such as pet dogs. This review describes the unique translational potential of the domestic cat as a natural model of AD, with reference to other animal models of AD.
Keywords
Alzheimer’s disease, pet cats, feline neuropathology, aging, feline cognitive dysfunction syndrome
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disease in humans, accounting for nearly two
thirds of dementia cases and affecting roughly 35 million
people worldwide [1]. The disease is characterized by
pathologic accumulations of two types of protein aggregates in specifc brain regions, which include plaques of
amyloid beta (Aβ) peptide in the neuropil, and neurofbrillary tangles (NFTs) composed of hyperphosphorylated
tau (pTau) protein found both within neurons and within
the neuropil as ghost tangles [2-3], but generally both of
these accumulations are thought to be necessary for progressive neuronal dysfunction, neuronal loss and cognitive
dysfunction in AD. The focus of pharmacologic trials has
shifted towards treating individuals in early stages of disease, with the goal to slow or prevent disease progression
and limit disability [3-4]. Genetically engineered rodent
models have been useful to identify candidate drugs for
early preclinical studies, however, candidate drugs validated in these models have generally been unsuccessful
in clinical trials [5]. Challenges translating therapies from
mice to humans may be explained by inherent differences
in neuroanatomy, lifespan, and physiology between the
species, as well as the fact that most mouse models result
from manipulation of one or more specific genes implicated in less common familial forms of the disease rather
than sporadic AD. Understanding the mechanisms and
pathogenesis in an animal model that naturally develops
neuropathologic lesions similar to AD is almost certainly
necessary for development of effective translational therapeutic strategies targeting early stages of the disease.
Several animal species have been shown to develop one
or more age-related lesions that are comparable to AD.
Ideally, animal models should possess 2 histopathologic
hallmarks of AD: Aβ peptide deposition (plaques) and
tau pathology (NFTs) [1]. In general, nonhuman primates
(NHPs) and dogs develop spontaneous Aβ deposition with
age, but do not reliably recapitulate tau pathology [6-7].
Wild-type rodents do not spontaneously form plaques or
NFTs, but genetically engineered mice have been developed that have single, double or multiple mutations
in genes responsible for the production of the Aβ and/or
tau proteins. Plaques are formed in these models by overexpression of mutant human amyloid precursor protein
(APP), resulting in overexpression of total Aβ beyond
physiologic levels or increasing the proportion of Aβ1-42,
which is more prone to pathogenic aggregation and has
a higher neurotoxicity profle comparted to Aβ1-40. NFTs
occur in some mouse models expressing mutations of
microtubule associated protein tau (MAPT), which causefrontotemporal dementia, not AD, in people. Although
helpful in preclinical trials, candidate drugs tested in
rodent transgenic models have very poor performance in
clinical trials [5]. By contrast, a limited number of studies
have shown that domestic cats can spontaneously develop
both histopathologic hallmarks of AD (Aβ deposition and
NFTs), as well as associated neuronal loss, in a pattern of
distribution similar to humans that progresses with age [5-
6] (Table 1). Additional studies involving large cohorts are
needed to further characterize neuropathologic similarities
between pet cats and humans and to correlate neuronal lesions with cognitive dysfunction.
Table1
Body and organ weights of Tg(TXN2)+/0 young mice .
| Study | Aβ | Tau | ||||||||
| Age | Preva lence | Distribution of plaques | Type of plaques | Distribution of poorly circumscribed deposits | Type | Age | Preva lence | Distribution | Type | |
| Nakamura et al., 1996 | >18y | 3/3 | cerebral cortex (temporal lobe > occipital lobe) | Aβ1-40 | cortical neuropil | Aβ1-40 | n/a | n/a | n/a | n/a |
| Brellou et al., 2005 | >17y | 4/4 | cortical layers of parietal lobe | strong Aβ1-42+, weaker Aβ1-40, Aβ8-17 | cortical layers of the frontal and parietal lobes | strong Aβ1- 42+, weaker Aβ1- 40, Aβ8-17 | n/a | n/a | n/a | n/a |
| Head et al., 2005 | >16y | 4/5 | prefrontal cortex, parahippocampal gyrus, parietal cortex, occipital cortex > outer molecular layer dentate gyrus | 4G8 and Aβ1-42 | prefrontal cortex, parahippocampal gyrus, parietal cortex, occipital cortex | 4G8 and Aβ1-42, infrequent Aβ1-16 (n=3/5) | >16y | 5/5 | hippocampus CA1, subiculum, and entorhinal/ parahimppocampal cortex in layers II and V | AT8+, anti-ubiquitin+ within dystrophic neurites (n=5/5); AT8+, PHF1+ within neurons (n=2/5) |
| Gunn-Moore et al, 2006 | >10y | 7/9 | cortical layers (deep) of anterior>mid-cerebrum | 4G8 | >11y | 2/9 | cerebrum, medulla, vestibular nuclei | AT8+ neurons | ||
| Takeuchi et al, 2008 | >10y | 6/6 | cerebral cortex and hippocampus | Aβ1-42 | cerebral cortex and hippocampus | Aβ1-42 | n/a | n/a | n/a | n/a |
| Chambers et al., 2015 | >8y | 14/15 | cerebral cortex | Aβ1-42 | intracellular oligomers in hippocampus | Aβ1-42 | >14y | 9/14 | entorhinal cortex, hippocampus > cerebellar cortex, locus coeruleus | AT8 and AT100 |
| Poncelot 2019 | >16y | 9/11 | temporal and frontalcortex | Aβ1-42+ | intracellular in hippocampus (n=1/9) | Aβ1-42 and Aβ1-40 | >18y | 4/6 | hippocampus and cortical subcortical structures | AT8 and PHF1 |
| Fiock et al., 2020 | >14y | 27/28 | Cortical layers IV and VI most prominent +/- hippocampus and basal ganglia | 6E10+ | >14y | 4/28 | entorhinal cortex > hippocampus, and neocortex | AT8+ | ||
Aβ plaques
Aβ plaques are derived from amyloid precursor protein
(APP), which is an integral membrane protein made by neurons and other brain cells, coded by the APP gene [3].
When APP is processed, it is cleaved at an extracellular
domain followed by an intracellular domain, and follows
one of two pathways. The non-amyloidogenic pathway
involves cleavage of the extracellular domain of APP by
an α-secretase, followed by intramembrous -secretase
cleavage, which prohibits the production of Aβ peptide.
Conversely, the amyloidogenic pathway involves cleavage of the extracellular domain of APP followed by intramembranous γ-secretase, which produces neurotoxic Aβ
peptides. The C terminus of A peptide is variable, ranging
from 36-43 amino acids in length, based on alterations
in the location of cleavage by γ-secretase [2]. In general,
the two dominant forms of Aβ in AD are Aβ1-40 and Aβ1-42.
Aβ1-42 is less soluble, more aggregation prone, and more
pathogenic than Aβ1-40 [1]. Once Aβ is produced, it accumulates in the extracellular spaces of the brain (parenchymal deposits) and may also be seen in vascular walls
(cerebral amyloid angiopathy, CAA). CAA can be seen
with, but is not exclusive to AD.
Parenchymal Aβ deposits may be classifed into subtypes
based on morphology, fbrils (B-sheet conformation), and
the surrounding elements (degenerative neurites [i.e. dystrophic neurites] and reactive astrocytes and microglia)
[8]. In general, there are two main subtypes: diffuse and
focal deposits [1-2]. The focal deposits are composed of
a focal spherical dense core of fbrillar Aβ (Aβ40 and Aβ1-
42) that stains positive for Congo red and thioflavin S. Focal deposits that contain few reactive glia and dystrophic
neurites (and thus lack a layered structure) are referred to
as primitive plaques. At a later stage, when they are centrally surrounded by more extensive dystrophic neurons
and reactive glia, they are referred to as mature or neuritic
plaques (NPs). The diffuse form of A deposits (DP) make
up the diffuse or amorphous plaque (DP), which are predominantly Aβ1-42. Morphologically they are large (>50
um), poorly limited, stain negative for Congo red and
Thioflavin S, are not surrounded by dystrophic neurites,
and typically do not have a surrounding glial reaction.
While both DPs and NPs can be found in normal aging,
the degree of cognitive impairment in AD patients is correlated with the severity of NPs, but not DPs. In addition
to routine H&E and special stains, Aβ deposits can also be
detected by IHC using antibodies such as Aβ1-16 (6E10),
Aβ8-17 (6F3D), and Aβ17-24 (4G8).
Topographically, parenchymal Aβ deposits are located
almost exclusively in the gray matter. The brain regions
involved depend on stage of disease, which can be classifed into fve successive phases (Thal Phases). The main
regions involved by phase include: neocortex in phase I;
hippocampus and entorhinal cortex in phase II; neostriatum and diencephalic nuclei in phase III; brainstem nuclei
in phase IV; cerebellum and additional brainstem nuclei
(pontine nuclei and locus coeruleus) in phase V [2, 5]. Deposition of plaques typically occurs frst as DPs, followed
by primitive plaques which progress to NPs.
In cats, Aβ deposition has been widely documented to increase with age, beginning by 7.5 years, and typically by
10 years of age [9-10]. Morphologically these are present
predominantly as DPs (Aβ1-42), and are negative for Congo
Red and thioflavin. They are most commonly found in the
cerebral cortex, with extension to the hippocampus and
basal ganglia. This is comparable to the distribution of
DPs in humans, which are most commonly found in the
cerebral cortex, basal ganglia, and cerebellar cortex [3].
Although typical NPs have not been identifed in cats, another form of Aβ deposition (intracellular Aβ oligomers)
has been found to accumulate within hippocampal pyramidal cells of cats 14 years and older [6]. The intracellular
Aβ oligomers were composed of hexamers and dodecomers, and found in the same brain regions as NFTs with
associated neuronal loss, similar to AD patients. Collectively, Aβ deposition in cats is most similar to early forms
present during normal aging and AD in humans.
Several other species have been shown to accumulate A
deposition with age including pet dogs and nonhuman primates (NHPs), in comparison to cats and humans (Figure
1). Similar to cats, both dogs and NHPs demonstrate agerelated deposition of Aβ the brain reminiscent of some but
not all features of AD. In dogs, the Aβ peptide is identical in sequence to humans, with deposits mainly as DPs
beginning at 8 years of age, composed primarily of Aβ1-
42 with minimal Aβ1-40, in similar topographic locations to
cats and humans [11-12]. Neuritic plaques of Aβ1-40 and
Aβ1-42 may occur less commonly, and mature NPs are very
rare in dogs [1]. In NHPs, age-related Aβ deposition has
been documented in great apes, old world monkeys, new
world monkeys, and prosimians, although there is a high
degree of variability between groups in the age of onset,
distribution, amount, and appearance of lesions. In general, DPs are most common across the groups, although
old world monkeys display more NP plaques compared to
other animals, beginning around 30 years of age [4, 7].
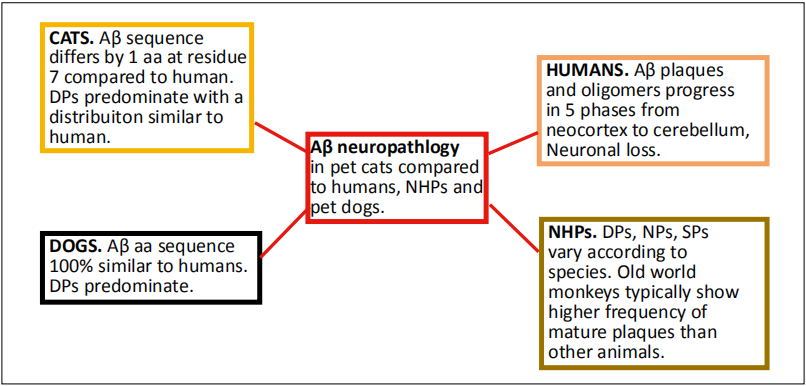
Figure 1. Summary of Aβ neuropathology in pet cats compared to humans, NHPs, and pet dogs. Abbreviations: NHPs- nonhuman primates; DP- dendritic plaques; NP- neuritic plaques; SP- senile plaques.
Tau aggregates (NFTs)
Tau is a microtubule-binding phosphoprotein in axons that
mediates axonal transport, encoded by the MAPT gene
[2-3]. Alternative splicing of MAPT yields a total of 6
isoforms in the human adult brain. Specifically, the isoforms differ by the number of amino acids inserted at the
N terminal aspect of the protein (0, 1 or 2 producing 0N,
1N or 2N) and by the presence of three or four amino acid
repeats (3R and 4R) in the C terminal part of the protein,
which contain the microtubule binding domains [13]. In
AD (a taupathy), tau is shifted from primarily axonal locations to a somatic-dendritic distribution, becomes hyperphosphorylated, ubiquinated, and loses its ability to bind
to microtubules, resulting in a misfolded, insoluble tau
protein. The specifc tau isoforms that accumulate in humans vary across different taupathies. pTau aggregates in
AD consist of hyperphosporylated 3R and 4R isoforms [2].
In addition, the active form glycogen synthase kinase-3
(GSK3), a tau protein kinase, has been found in association with tau lesions and at increased expression levels
in AD brains [13]. Tau aggregates may be present in the
neuron cell body, dendrites, and axons. In the cell body,
tau aggregates progress from pretangle lesions (diffuse or
granular phospho-tau immunoreactivity in the absence of
fbrillary structure) then become fbrillary, forming neurofbrillary tangles (NFTs). NFTs that form in the neuronal
cytoplasm result in neuronal degeneration and death, after
which the NFTs persist in the neuropil as ghost tangles.
Tau aggregates may also accumulate in dendrites, where
they are known as neuropil threads. Lastly, tau aggregates
can be present in axons, where they are visualized as fne
tau-positive processes that make up the neuritic component (crown) of the focal plaque.
Tau aggregates are usually detected by IHC using immunostaining with anti-tau antibodies such as AT8, PHF1,
and anti-3R and anti-4R. Special stains such as silver
stains and thioflavin S can also be used [1]. Ultrastructurally, tau aggregates in AD are made of two filaments
paired in a helical structure (PHFs). Straight filaments
have also been seen, but usually these are more suggestive
of other taupathies such as progressive supranuclear palsy
(PSP) and Pick’s disease (PiD) [13]. In general, the development of Aβ plaques is considered to precede and accentuate the develop of NFTs in human AD [14]. The burden
of tau pathology progresses from medial temporal lobe
to neocortex describred through Braak stages, as detected
by AT8 immunohistochemistry. The stages sequentially
involve the medial temporal cortex (stage I), hippocampus
CA1 (stage II), subiculum and other regions of hippocampus and medial temporal cortex (stage III and IV) and
other areas of neocortex (stage V and VI) [2].
Aging cats have been shown to develop tauopathies that
share many features to human AD. For example, adult
cats express 6 total tau isoforms, including the hyperphosphorylated 3R and 4R isoforms, which can form aggregates in the presence of Aβ as in human AD [6, 13].
Cats also show increased expression of GSK3 (tau protein
kinase) in association with tauopathies and regions of
neuronal loss at 15 years of age [15]. Morphologically,
neurons with intracellular phosphorylated tau in aging cats
have been noted to display a sprouting response similar to
human AD [15]. Several forms of tau aggregates reported
in cats include: pretangles, threads, dystrophic neurites,
NFTs and ghost tangles [16]. Ultrastructurally, cat NFTs
are similar to those in humans, consisting of some straight
flaments but mostly paired twisted patterns of flaments.
These features of feline NFTs accompanied by neuronal
loss and occurring in intracellular oligomers in the same
brain region (hippocampus), have also been described in
transgenic mouse models and human AD patients, and
thus represent a potentially high impact naturally occurring model of the disease [5] compared to pet dogs and
NHP’s (Figure 2).
Although most other aging nonhuman animals do not
spontaneously develop NFTs and neuronal loss, abnormal
tau phosphorylation has been described in a number of
species including sheep and goats, bison, bears, degu, and
wolverines [1, 13]. Dogs have not been shown to develop
NFTs, but do show increased phorphorylation of tau that
may represent pretangle pathology. In NHPs, NFTs are
relatively rare, and when present are usually focal, mild,
and not associated with neuronal loss [4, 7].
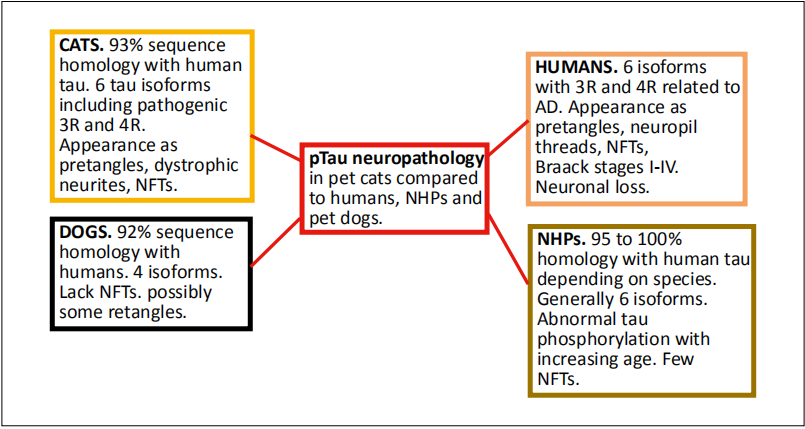
Figure 2. Summary of pTau neuropathology in pet cats compared to humans, NHPs, and pet dogs. Abbreviations: NHPs- nonhuman primates; NFT- neurofbrillary tangles.
Cognitve dysfuncton
Recent clinical and research criteria have identifed three
progressive stages of human AD: preclinical, prodromal,
and dementia [2]. During the preclinical period, there are
no symptoms of cognitive dysfunction. However, neuropathology associated with AD (accumulation of Aβ deposition followed by tau pathology and neuronal degeneration/synaptic dysfunction) occurs during this period and
may precede initial symptoms by decades [14]. Clinical
signs of mild cognitive impairment occur during the prodromal stage followed by progression to dementia (clinical
diagnosis of AD) based on cognitive-behavioral status.
Typically, AD presents as an initial decline in episodic
memory, followed by progressive decline across multiple
cognitive functions or intellectual symptoms (ex: amnesia,
aphasia, apraxia, agnosia) and behavioral changes (personality changes, depression, hallucinations, delusions) that
impair social function and difficulties with daily living.
Therefore, clinical AD is considered a late stage of disease
progression, and biomarkers characterizing preclinical and
prodromal stages are essential for interventions aimed at
preventing or reversing disease progression.
The association between age-related neurodegenerative
lesions and cognitive dysfunction in cats is not as well
described as for humans or dogs. However, both dogs and
cats can develop age-related deterioration in cognitive
functions manifested as behavioral changes that are similar to cognitive domains affected in AD [14]. Cognitive
dysfunction syndrome (CDS) is the term used to describe cognitive decline in geriatric cats and dogs after excluding
underlying causes of illness or behavior effects of drugs.
CDS is estimated to affect roughly one third (28 percent)
of cats 11-14 years old, and 50 percent of cats 15 years of
age or older [19]. The incidence of CDS is similar in clinical settings in dogs, which is estimated to affect roughly
28 percent of dogs 11-12 years old, and 68 percent of dogs
over 15 years old (although lifespan and biological age in
dogs varies by breed) [9, 17]. Human dementia displays
similar trends, affecting roughly 1-3 percent of people 65-
70 years old and 50 percent of people over 85 years of
age. In cats, there is not yet a validated relationship between cognitive/behavioral dysfunction and extent of Aβ
deposition [15], but aged cats that display signs of behavioral dysfunction have been found to have Aβ plaques [16,18]. An association between tau pathology and CDS also
remains to be more clearly clarifed, but several studies in
aged cats showed that all cats affected with tau pathology
(4/4 cats >15y and 5/5 cats 16-21y) displayed changes
in behavior suggestive of cognitive/memory impairment
[11, 13]. The pattern of Aβ deposition in dogs has been
documented to increase with age, and extent of deposition is positively correlated with cognitive impairment [9-
10]. Although dogs do not develop NFTs as in humans
with AD, a study of multiple breeds of dogs demonstrated
that intracellular phosphorylated tau (Ser396) expression
in neurons and astrocytes of the cerebral cortex and hippocamus was associated with cognitive dysfunction. Agerelated defcits in memory and attention, deposits of amyloid plaques, and atrophy and/or loss of cholinergic and
monoaminergic neurons are well documented in NHP [4].
Further studies of larger cohorts are needed to expand
on the neuropathologic and cognitive changes that have
been characterized thus far in aging cats. Studies could
be designed that borrow from canine studies in which
samples are obtained from clinical settings, while also
addressing issues identifed in feline studies including behavioral changes relevant or more common in feline CDS
compared to dogs. For example, archived brain samples
from potentially large cohorts could be accessed from
veterinary clinics retrospectively from young and old cats
that were euthanized or died spontaneously from various
disorders, excluding those with major brain lesions such
as neoplasia or inflammatory disease evident grossly or
microscopically. Corresponding clinical records could be
subjected to a scoring system to group cats into cohorts
of cognitive decline (present/absent) or adapted/modified from one of the several available scoring systems
for screening severity of age-related cognitive disorders
in pet cats (ex: absent, mild, moderate severe). Available
clinical and historical data to evaluate for the presence of
concurrent disease would include physical exam fndings,
laboratory test results including bloodwork and urinalysis,
results of screening for infectious disease such as FeLV
and FIV, imaging and medications [14-16]. Examples of
common diseases that may impact behavior in cats (and
people) include hyperthyroidism, chronic renal disease,
hypertension, osteoarthritis and diabetes mellitus [17, 19].
Ideally, with a large enough cohort, these diseases could
be randomized between groups of cats with cognitive
dysfunction and those without. Collectively, studies that
incorporate these elements could help to better address the
question of whether neuropathologic lesions of AD correlate with clinical signs of behavioral/cognitive dysfunction, and whether there are any effects of comorbidies and
breed on the extent of neurolopathologic changes.
Conclusion
In summary, A deposition and tauopathy (including NFT formation) with neuronal loss can spontaneously develop in aging pet cats in a similar distribution and with similar characteristics to human AD. Other nonhuman mammalian animals rarely develop all three of the major pathologies that characterize AD. Concurrent with the presence of neuropathologic lesions, cats have been reported to develop cognitive impairment with increasing age; however, the correlation with neuronal lesions is not yet fully validated in this species. The presence of AD-like neuropathology positions the aging pet cat as a potentially high impact model to investigate pathogenesis and therapeutic interventions for AD with potentially superior translational and preclinical predictive power compared to pet dogs and nonhuman primates. However, additional research on large cohorts of pet cats is needed to confrm these exciting and novel but preliminary observations.
Declarations
Authors’ contributions
Jenna Klug wrote the first draft. All co-authors contributed to editing and additions. Warren Ladiges did the final editing.
Conflicts of interest
The authors declare no conflicts of interest.
Consent for publication
All authors consent to the publication of this manuscript.
References
1. Youssef S A, Capucchio M T, Rofina J E, et al. Pathology of the aging brain in domestic and laboratory animals, and animal models of human neurodegenerative diseases. Veterinary pathology, 2016, 53(2): 327-348.
2. Calderon-Garcidueñas A L, Duyckaerts C. Alzheimer disease. Handbook of clinical neurology. Elsevier, 2018, 145: 325-337.
3. Kumar V, Abbas A K, Fausto N, et al. Robbins and Cotran pathologic basis of disease, professional edition e-book. Elsevier health sciences, 2014.
4. Emborg M E. Nonhuman primate models of neurodegenerative disorders. ILAR journal, 2017, 58(2): 190-201.
5. Fiock K L, Smith J D, Crary J F, et al. β-amyloid and tau pathology in the aging feline brain. Journal of Comparative Neurology, 2020, 528(1): 112-117.
6. Chambers J K, Tokuda T, Uchida K, et al. The domestic cat as a natural animal model of Alzheimer’s disease. Acta neuropathologica communications, 2015, 3(1): 78.
7. Heuer E, F Rosen R, Cintron A, et al. Nonhuman primate models of Alzheimer-like cerebral proteopathy. Current pharmaceutical design, 2012, 18(8): 1159-1169.
8. Brellou G, Vlemmas I, Lekkas S, et al. Immunohistochemical investigation of amyloid ß-protein (Aß) in the brain of aged cats. Histology and histopathology, 2005.
9. Vite C H, Head E. Aging in the canine and feline brain. Veterinary Clinics: Small Animal Practice, 2014, 44(6): 1113-1129.
10. Yu C H, Song G S, Yhee J Y, et al. Histopathological and immunohistochemical comparison of the brain of human patients with Alzheimer’s disease and the brain of aged dogs with cognitive dysfunction. Journal of comparative pathology, 2011, 145(1): 45-58.
11. Chambers J K, Mutsuga M, Uchida K, et al. Characterization of AβpN3 deposition in the brains of dogs of various ages and other animal species. Amyloid, 2011, 18(2): 63-71.
12. Takeuchi Y, Uetsuka K, Murayama M, et al. Complementary distributions of amyloid-β and neprilysin in the brains of dogs and cats. Veterinary pathology, 2008, 45(4): 455- 466.
13. Poncelet L, Ando K, Vergara C, et al. A 4R tauopathy develops without amyloid deposits in aged cat brains. Neurobiology of aging, 2019, 81: 200-212.
14. M, Nichol J, Araujo J A. Cognitive dysfunction syndrome: a disease of canine and feline brain aging. Veterinary Clinics: Small Animal Practice, 2012, 42(4): 749-768.
15. Head E, Moffat K, Das P, et al. β-Amyloid deposition and tau phosphorylation in clinically characterized aged cats. Neurobiology of aging, 2005, 26(5): 749-763.
16. Gunn-Moore D A, McVee J, Bradshaw J M, et al. Ageing changes in cat brains demonstrated by β-amyloid and AT8-immunoreactive phosphorylated tau deposits. Journal of feline medicine and surgery, 2006, 8(4): 234-242.
17. Gunn-Moore D, Moffat K, Christie L A, et al. Cognitive dysfunction and the neurobiology of ageing in cats. Journal of Small Animal Practice, 2007, 48(10): 546-553.
18. Nakamura S, Nakayama H, Kiatipattanasakul W, et al. Senile plaques in very aged cats. Acta neuropathologica, 1996, 91(4): 437-439.
19. Gunn-Moore D A. Cognitive dysfunction in cats: clinical assessment and management. Topics in companion animal medicine, 2011, 26(1): 17-24.