Open Access | Review
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
The cellular senescence unifcation model and telomerase therapy: To treat all age-related diseases
* Corresponding author: Steve Liebich
Mailing address: Department of Biomolecular Science & Chemistry, Clarkson University, 10 Clarkson Ave, Potsdam, NY 13676,
USA.
E-mail: liebicsf@clarkson.edu
Received: 07 June 2020 / Accepted: 18 Septemper 2020
DOI: 10.31491/APT.2020.09.030
Abstract
Since the discovery of the telomere by Hermann Muller and Barbara McClintock in years 1938-1940, a great progress has been made in molecular genetics and in the relatively new field, biogerontology. Almost 40 years have passed since the discovery of telomerase by Carol Greider and Elizabeth Blackburn (1984). Since those major discoveries, the scientific community has linked many naturally occurring ageing mechanisms in cells to the shortening of telomeres and the lack of telomerase activity in these cells. A great number of mutagens, radiation, and toxic chemicals negatively impact the length of telomeres with their truncation triggering a fatal cascade of events inside the cell which can lead to the state of senescence and eventually to cell death. Even though cellular and bodily ageing is a complex, multilevel, and highly orchestrated natural process happening at the subcellular level of all multicellular organisms, and a single known mechanism is extremely insufficient to explain all observed molecular and morphological changes, a unified cohesive theory of ageing must exist. The cellular senescence unification (CSU) model, combining the free-radical-mitochondrial and telomeric theories, helps establish a strengthened base for future therapies of all age-related disorders. If the CSU model is strong enough to explain and describe most of all the observed alterations in the ageing cell, a focused and deliberate therapy might be developed. This work introduces telomerase therapy as an efficient, highly effective, and clinically favored treatment for most age-related disorders, which can be elucidated by the CSU model. Gene therapy is the next natural step forward for biogerontology since the discovery of the telomere 80 years ago.
Keywords
Telomeres, telomerase therapy, cellular senescence, unified theory of ageing
Introduction
Each eukaryotic cell division leads to a gradual sequence
loss at the chromosomal termini known as telomeres [1].
This so-called end replication problem (ERP) forms the
basis of cellular senescence, along with a few other wellestablished biochemical processes disrupting the cellular
homeostasis [2].
Telomeres are composed of repeated oligonucleotide sequences supported by a six-protein complex (shelterin)
and the quaternary structure they form in vivo [3-5]. Thus,
telomeres are the specific genomic protection device, guarding the cell from the inexpedient recombination
events, degradation through the DNA repair system, and
signifcant genetic material loss [6-9]. In some cell types,
a reverse transcriptase known as telomerase reconstitutes
the original length of the shortening chromosomes, saving
the chromosomal and genomic stability of the cell [10-14].
In such conditions, the cell undergoes almost an infinite
number of mitotic cycles, breaking through the Hayflick
phenomenon [15].
Cellular and bodily ageing result from complex, multilevel, and precisely controlled mechanism of decline in effectiveness of physiological processes in the cell and decline
in the stability of its genome. Because of this complexity,
more than three hundred different or slightly different
theories of ageing have been proposed [16]. Many the
ories overlap, while the others leave gaps too broad to be
neglected. New data are very often misleading and contradicting, thus supporting neither of the proposed rationales. Therefore, biogerontology needs to fnd a common
denominator for all credible theories, so that one elegant
unifed theory could explain the entire life-long process of
cellular ageing.
All proposed ageing theories are segregated into two main
categories: programmed and non-programmed [17]. The
frst category takes into account all ageing factors genetically inherited and gradually manifested over the span of
a lifetime. The second group includes theories emerging
from the customarily occurring errors in the genome and
accumulated damage in the cell; tear-and-wear is preferably chosen as the classic theory considering accumulating damage in the genomic DNA. However, it had been
the free-radical and mitochondrial theories that took much
of the scientifc community’s appraisal in the last century
[18-21]. Three decades ago, the telomeric theory of cellular ageing stole the spotlight and since has grown into
a well-developed and data-supported biogerontological
doctrine, which aims to explain most of the observed hallmarks of cellular senescence [22-25]. These two theories,
the free radical-mitochondrial and telomeric, have the
potential to form a single comprehensive model defining the complexity of ageing. It is referred to here as the
cellular senescence unifcation (CSU) model. For clarity
purposes and due to space limitation, only the telomeric
theory will be introduced and described.Is telomerase, the
enzyme of capacity to restore the shortened telomeres and
stabilize the genome, the defnitive answer to the puzzling
problem of ageing? Could we take advantage of this fnding and apply it to humans, hence aim to improve health,
fight age-related diseases (ARDs), and even reverse the
ageing machinery in their cells? This article elaborates on
the subject and aims to address these two questions with
proper and preservative analysis of recent advances in the
feld of biogerontology.
The cellular senescence and ageing unifcaton theory
As Bodnar et al. showed, each human cell which does not
express an active hTERT (human telomerase reverse transcriptase) transcript loses its terminal chromosome repeats
[11]. If cellular senescence was viewed as a dense system
of all cellular and genomic changes occurring over an undefned span of time, relative loss of telomeres would be
both one of the causes and effects of the total intracellular
changes. By now, telomere loss and lack of telomerase expression in ageing cells are some of the most studied phenomena in ageing biology. However, it is not, and it must
not be treated as the cause of gradual cellular dysfunction.
Since the paper of L. Hayflick and P. Moorhead on the
limited mitotic capacity of somatic cells was published,
the link between the restricted cell doublings and their
mortality, telomere shortening and the inevitable replicative senescence phenomenon has become obvious [15]. A
large number of publications have been dedicated to the
association of telomere shortening with numerous agerelated disorders (see below).
A few aspects of cellular senescence must be considered
before further discussion. Senescence is not equivalent
to the quiescent state of the cell [26]; cell lines like hepatocytes and corneal endothelial cells maintain replicative
capability, but do not divide without external stimuli [27,
28]. Cellular senescence is defned then as the permanent
quiescence of the cell cycle. The all-or-nothing model of
cellular senescence, popularized in the past century, has
lost its merits over the more consistent and data-supported
gradual changes within the cells [22]. Today, the cell senescence model of aging is acknowledged and the postulate that senescence and the non-programmed errors accumulated in genome result from changes in, among others,
gene expression and the epigenetic landscape seems to be
satisfying and unify other related theories [29].
Due to space limitation and deliberate focus on the telomeric theory, some programmed and non-programmed
factors of cellular ageing further explained will be discussed in the light of telomeres shortening, however, those
factors reach beyond the telomere biology and affect a
plethora of other intracellular sites and processes. Recent
research advances in biogerontology show a moderately
strong correlation between telomere shortening and other
possible processes contributing to cellular ageing, which
all cumulatively (and defnitely not singlehandedly) lead
to the senescent state of the cell, which can be defned as
a quiescent (non-mitotic) state of the cell unresponsive
to the external stimulation and such cell is found in the
G0S stage of the cell cycle. As discussed below, telomere
shortening, chromatin destabilization, change in genome
dynamics, uneven generation of free radicals, and alteration in gene expression
patterning are unequivocally correlated, which may help to draw more defnitive conclusions in the future (Figure 1).
After all, senescence is a gradual process occurring in
every cell of a multicellular organism, including cells that
are mitotically inactive. Senescence should be understood
as a complex network of changes in genes expression,
DNA damage, ineffective DNA repair mechanisms, misbalance between reactive oxidative species production
and scavenging, cell morphology, toxins accumulations,
proteomic changes, and finally loss of telomeres. The
telomeres feld has had over half a century of research and
promising findings which in consequence makes it both
one of the most studied cellular senescence biomarker
and the target for potential gene therapy; the encouraging
studies of re-lengthening the shortened telomeres in animals (in vivo and in vitro) and in humans (in vitro) only
seal the inevitable importance of the telomeres role in
ageing. Even though it is subjective and experimentally
unapproachable to test whether a cell is already senescent
or “not yet,” the CSU model is supported by our current
knowledge, and although not perfect, it is still the best tool
we can use to not only explain cellular ageing processes,
but also to manipulate them.
Fossel (2012) suggests the relative telomeres length measures as a reliable, clinically practical, and specific biomarker of age-related diseases [30]. Correlation between
the telomere corrosion in a chronologically old individual
and onset of various age-related diseases is well-known,
thus telomeres and their shortening seem to be a reliable source of clinical information and a platform for proper
intervention. It must be noted that it is the relative rate
of telomeres shortening, not just a total loss of their sequences. This gives an insight into the onset of telomeredependent diseases and their clinical manifestation [31].
This remark has been taken to clinics with commercial
enterprises of key telomere researchers: Maria Blasco (Life
Length) and Calvin Harley & Elizabeth Blackburn (Telome
Health). These are only the frst steps of the telomere research evolving into clinical importance.
However, as long as the relative telomere loss is a strong
biomarker in chronologically advanced patients and individuals in greater risk cohorts, the very common telomere
length (TL) measure in peripheral blood leukocytes (PBLs)
seem to be an undesirable method. First, false negative
results might affect the diagnosis of a patient still affected
by slowly evolving pathological processes. The reason is
that leukocyte telomeres might remain relatively stable if a disorder develops in the liver, for instance; moreover,
the “old” leukocytes are constantly being replaced by the
white blood cells with long telomeres. Second, peripheral
leukocytes can be exposed to toxic, stressful or immunological factors that would influence telomere loss in the
PBL without any underlying age-related disorder [32].
Multiple studies showed a positive correlation between
external factors such as smoking, obesity [33], oxidative
damage [34, 35] and past infectious diseases [36-38].
Third, the genomic changes of white blood cells (WBC)
are just as important as in other human cells: the age-dependent telomere shortening [39], polymorphisms in the
hTERT promoter and its regulatory genes [40] (although
a Swedish cohort studies of the same single nucleotide
polymorphism did not show any correlation [41], and
changes in the epigenetic landscape of the hTERT [42].
These findings indicate that the relative PBL telomere
loss is a signifcant biomarker for a number of age-related diseases, but PBLs are not always the right source of telomere attrition information.
Coronary heart disease [43], osteoporosis [44], diabetes
[45], and other age-related disorders are correlated with
shortened telomeres; the telomere attrition leads to cell senescence as observed in multiple tissues and organs. PBL
telomere length (TL) measurements have been applied
to a plethora of various age-related disorders, diseases of
affluence and immunological disorders, including longitudinal studies of cardiovascular health problems [46],
chronic obstructive pulmonary disease [47], familial and
sporadic pulmonary fbrosis [48], and hematopoietic malignancies [49]. These data underlie a strong correlation
between telomere attrition and the etiopathophysiology of
diseases observed in clinical settings.
Human skin fbroblasts were the frst telomere-associated
senescence model cells, for which the telomerase transfection proved to be liable [11]. In two parallel studies,
human keratinocytes and fibroblasts were grown on an
immune-compromised mouse and the new skin morphology was assessed for early (20 population doublings) and
late (85 population doublings) passages; the late passage
cells were further transformed with an external hTERT
and the re-lengthened telomeres led to skin reconstitution,
optimal gene expression and normal flamentous connections observed in the young skin [50].
Almost every tissue type in the human body demonstrates
histological and ultracellular changes associated with telomere shortening. For cardiovascular diseases, mice and
human models show exceptional correlation between telomere dysfunction (also oxidative stress, proinflammatory
molecules activation, etc.) and senescence of vascular endothelial cells leading to development of cardiomyopathy
and severe atherosclerosis [51]. The impairment of control
mechanisms of stem cell reserves and their differentiation
and division in the bone marrow, hugely associated with
telomere attrition and immune system age-related changes, is responsible for the pathological decrease in activity
in older people. Similar deferment in physiology has been
found in glial cells, the cells that divide and proliferate in
the brain, whose ultracellular ageing transformation has
been presented to be the leading cause of Alzheimer’s disease [52]. These examples indicate the necessity to reform
the way how one interprets the pathophysiology of many
age-related disorders. The non-divisional pattern of neural
or myocardial cells would lead to an inaccurate and misguided conclusion that the telomeric theory could not be
taken into account for these cells. However, this inaccuracy stems from the wrong perspective: one must shift focus
from the mitotically inactive to the dividing cells found in
the proximity of the non-divisional cells. The combined
pathophysiologic outputs of the non-dividing, but still
homeostatically deteriorating cell and the mitotically active cell, whose telomeres do truncate after each round
of mitosis (next to other countless ultracellular ageing
processes) draw a more consistent and self-explanatory
picture of age-related disorders.
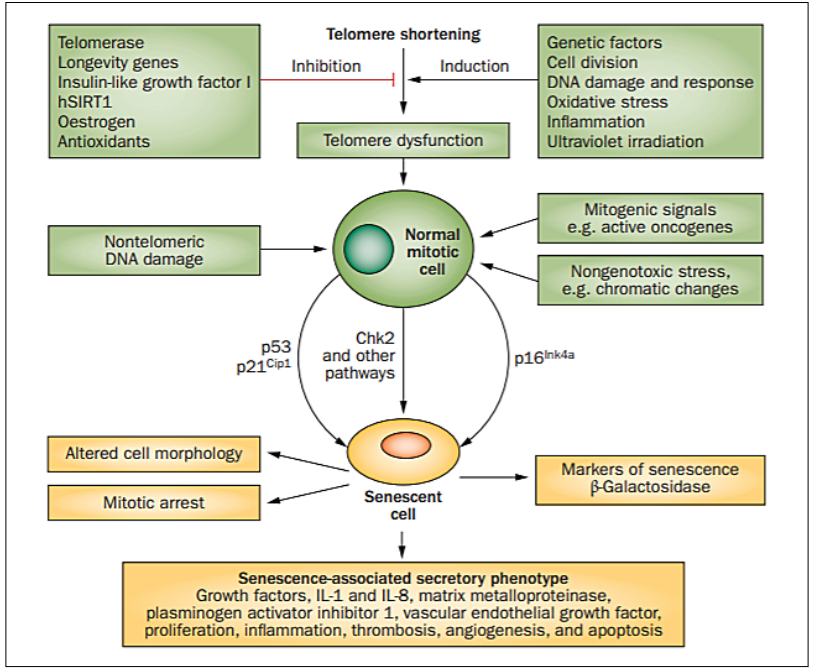
Figure 1. Cellular senescence: external and internal factors. A plethora of mutagens, signaling pathways, cytokines, and oxidative agents influence the rate of telomere shortening in the cell. Environmental stressors (UV light, industrial toxins, carcinogens, and intercalating agents), expressed oncogenes, and chromatin alterations also affect the progression of cellular senescence. Tumor suppressors, including p53, are activated upon those deleterious signals and respond adequately by triggering mitotic arrest, altering cell morphology, secretion of growth factors, cytokines, and apoptotic factors. Telomeres are affected by all aforementioned senescence factors, but the same factors induce their effects through other genomic and non-telomeric pathways. Abbreviations: hSIRT1 (NAD-dependent protein deacetylase sirtuin-1); Chk2 (checkpoint signaling kinase 2). Figure adapted from [51].
Control mechanisms of telomere length and telomerase expression
Senescence is a gradual process that takes place in every living cell of the human body, with cells varying in
degrees of senescence. This is a complex and global process incorporating changes in epigenetic status, genomic
mutations and breakages, slowed metabolic turnover of
the cell, increased levels of oxidative species, failing mitochondria, accumulation of toxic chemical compounds
(external and internal), deterioration of signaling pathways, increased turnover of proteins, and increasing area
to volume ratio of the cell.
Not all changes lead to the cell’s death though. Senescent cells rarely “die,” they rather move to a quiescent
state where they become less or completely mitotically
inactive. However, shortened telomeres in somatic cells
advance genomic defects, and this destabilization leads to
more rapid cellular changes [23, 25]. On the other hand,
stem cells are partially protected from the instability by
expressing telomerase, an enzyme that extends the missing chromosomal termini. The difference between somatic
and stem cells is that the former has an accelerated rate
of senescence, while the latter gradually become more
senescent [53, 54]. Thereby with old age, stem cells are
more senescent than they were in the young organism, but
somatic cells are more senescent than the stem cell populations. While most organs store their somatic stem cells
in so-called niches, which function as “spare parts” for
the ageing organ, the process of replacing the aged and/or
dead somatic cells with transit cells (dividing and slightly
differentiating stem cells) demonstrating self-renewal capability diminishes [54-56].
Telomerase plays an important part in keeping the cell’s
homeostasis balanced. Therefore, telomerase expression
and activity regulation in the cell is highly significant.
The 40-kb hTERT locus sits on chromosome 5, only 2 Mb
away from the chromosomal end [57]. hTERT lacks both
TATA and CAAT boxes, but is regulated with two canonical E-box sequences [12] (Figure 2). The E-box sequences
and numerous CpG islands provide large methylation sites
for the hTERT locus, which demonstrates high epigenetic
regulation [58]. Most human cells do not produce telomerase [12]. The exceptions are stem cells, germ cells, and
select white blood cells [59, 60]. Moreover, 85 – 90% of
cancer cells express active telomerase [61-63]. It is still
unknown why most somatic cells are hypermethylated at
the telomerase site and what exact mechanisms participate
in the negative regulation of the locus [64]. However,
various fndings show that some cells are capable of producing telomerase in vivo, but these are inactively spliced
variants that do not contribute to the telomerase activity
[65].
The telomerase-telomere cellular “apparatus” is also controlled by a wide network of proteins. The trans-acting
regulatory proteins are extremely active at the hTERT
locus. The most abundant transcription factor (TF) complexes are E-box-binding c-Myc/Max complex, which cooperates with the GC-box far-flung Sp1 TF acting as the
activating element [66]; on the other hand, Mad1/Max
complex, along with deacetylases, represses the hTERT
locus [67]. To review the divergent transcription factors
involved in the telomerase expression regulatory mechanisms refer to [68-70].
Telomeres are supported by another, but not exclusive,
set of proteins. The already mentioned shelterin complex, composed of six core proteins, both protects telomeres from exonucleases degradation and provides an
interaction matrix for other regulatory factors [71]. The
telomeres instability is reflected mainly through the double-strand break (DSB) events. The DSBs induce chromosomal translocations, error-prone recombination events
and tumorigenic processes [72]. Two major DNA damage
sensing pathways are related to ATM and ATR serinethreonine kinases, which are activated at cell cycle checkpoints, mostly at G0, G1 and S; the DSBs are repaired by
two main processes: homologous recombination (HR) and
non-homologous recombination end-joining (NHEJ) [73].
A plethora of proteins are used by the cell to protect the genomic stability and the truncated telomeres through the
two recombination mechanisms [73, 74].
This complicated and heterogeneous network of signaling
pathways and trans-acting regulatory elements serves to
maintain telomere length and respond to occurring changes at telomeres. Recently, the telomere position effect
(TPE), imposed by relative shortening of telomeres and
the rate of loss affecting nearby genes through telomeric
chromatin looping has attracted much attention [75, 76].
This is because the TPE explains how physical changes
of the truncated telomeres affect other gene expression
through physical mechanisms, very often affecting genes
placed more than 10 Mb away from the telomeric region
[75]. Additionally, short telomeres themselves may serve
as another proximate senescence trigger through DNA repair mechanisms [77]. However, many researchers agree
that, apart from the aforementioned mechanisms, the relative length of telomeres and the rate of telomere loss affect
the genomic structure at most [30, 53, 78, 79]. The cell
responds accordingly to all these changes, and depending
on its current biochemical status, becomes more receptive
to other deleterious processes inside of it.
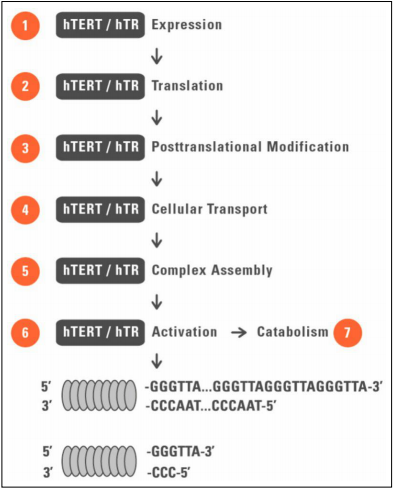
Figure 2. Telomerase expression mechanism in telomerase-positive cell line. Telomerase is a ribonucleoprotein complex composed of many subunits, of which telomerase reverse transcriptase (TERT) and telomerase RNA component (TERC) are essential in physiological activity of telomerase. The multicomponent enzymatic complex extends chromosomal termini by synthetizing de novo tandem telomeric repeats (5’TTAGGG-3’) at 3’ end of the chromosome. TERC binds the 5’ C-rich strand and its 11-nt sequence serves as the replication template for reverse transcription. Abbreviations: hTERT (human telomerase reverse transcriptase); hTR (human telomerase RNA component). Figure adapted from [115]..
The role of telomerase in cellular pathophysiology
The negative correlation between the telomerase expression and the onset of most cancers in the human body has
been favored by the scientific community for years [61-
63]. The cancerous cells’ replicative immortality stressed
by Hanahan and Weinberg has been associated with the
hTERT locus activation, and thus the conclusion withdrawn was simple: telomerase expression promotes tumorigenesis in somatic cells [80].
Interestingly, the remaining cancer types that do not
activate the hTERT locus still elongate their telomeres
through the alternative lengthening of telomeres (ALT)
mechanism [81, 82]. This route of telomere elongation
takes advantage of chromosomal instability and strands
breaks, which is detrimental for the cell karyotype. A tumorigenic clone line will always sort the cells with longer
telomeres compensating the higher proliferative rate [81];
furthermore, the telomere sister chromatic exchange (TSCE), telomere-repeat arrays exchange resulting from the
ALT pathways, yields the unequal sister telomere lengths
in different cancerous cell lines [83].
The misconception is that long telomeres are a biomarker
for high cancer risk; this is far from the truth. In fact, in
most cancerous cells the telomeres are much shorter than
those found in normal cells [84, 85]. Most recent tumor
studies fnd that telomeres in the cancer cells are, through
structural analysis, truncated and recombinogenic: dicentric chromosomes, reciprocal translocations and other
deleterious karyotypes [86, 87]. Moreover, in most studies
correlating the telomere length with concomitant cancer, the telomere length has either been found normal or
reduced. In other studies, the telomere length has been found increased with either active or quiescent telomerase
[88-91]. In many papers studying the correlation between
telomeres length, telomerase expression and cancer cell
biology, the histopathological changes in cancer tissue
are not reported. Not only is the average length of the
cell telomeres measured instead of the shortest telomere
length, but these telomere length measurements are taken
from the patient’s PBLs suffering from a malignant disease that is not correlated with the immune system [30].
These mistakes generate false conclusions and consolidate
the misconception of the negative correlation between the
telomerase expression and tumorigenesis.
There is much evidence on the relationship between the
telomere length maintenance (TLM) and cancer. However, there are no data indicating that cancer is caused
by the overexpression of telomerase or the overly long
telomeres. Present studies with somatic cells expressing a
recombinant telomerase gene or somatic cells treated with
telomerase vectors, and studies on the embryonic pluripotent cells with constitutive telomerase expression, indicate
no cancer remarks [26, 92-94]. This can be partially explained by the fact that only the average telomere shortening and its rate, not the total telomere loss, determine
the senescence phenotype; thus telomerase expression increases genomic stability by keeping the average telomere
loss and re-lengthening constant [95]. Simultaneously, the
number of cell divisions increases (uncontrolled growth),
which leads to further telomere truncation which must
be overcome by a higher rate of telomerase expression
(Figure 3). These findings can be supported by showing that knocking down the telomerase gene coincides with
tumor growth arrest, but only when the tumor had already
grown, not before the tumorigenesis process, not even in a
p53-null cell [79].
However, telomerase expression does not serve its canonical function only. Recent fndings present multiple non-canonical functions of telomerase, which influence genomic
stability, very often resembling the ones found in the ALT
mechanisms [96]. All in all, both the telomerase- and
ALT-based telomere length maintenance pathways stabilize the already destabilized genome of the cancerous cell.
Thereby, it is not about a maximum telomere length that
can keep the cancer cell from apoptosis, but about keeping telomere length above the critical threshold, different
for each organism. This also explains why tumor cells in
mice upregulate the mTERT expression, even though the
mural telomeres (approx. 50 kb) are much longer than the
human’s (approx. 15 kb-long in a young human cell) [78].
This notion does not hold true for some individuals, however, if their cellular environment manifests a variety of
mutations (genome) and preconditioned disorderliness in
biochemical pathways and DNA repair mechanisms (cell);
although in these cases the telomerase expression introduces new level of complexity to the unbalanced system
rather than becoming the leading cause of tumorigenesis.
The studies in mice corroborate the observations that,
contrary to the general assumptions, telomerase does not
increase the risk of tumor development, but also protects
the cells from cancerogenesis [97, 98].
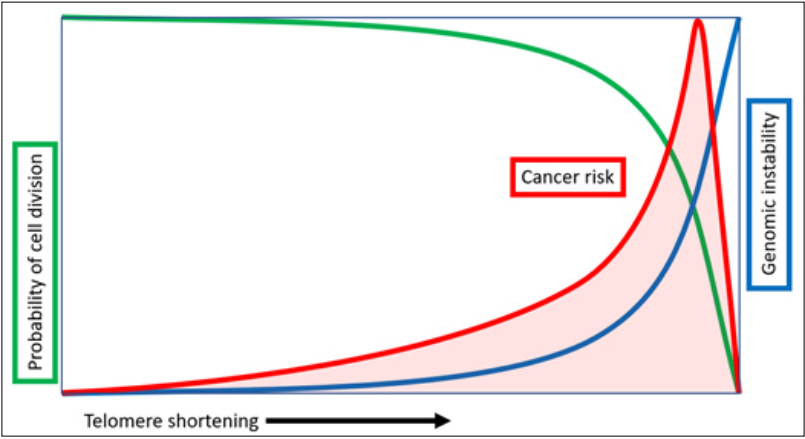
Figure 3. Relationship between telomere shortening, increasing genomic instability and varying cancer risk. With each replicative division the total telomere length decreases, which leads to miscellaneous genomic and intracellular alterations increasing the overall risk of hyperproliferation and tumorigenic mechanisms development. It must be noted that a cell in order to develop cancerous changes must rely on relatively long telomeres for proper genomic stability. If its telomeres are too short, the cell drifts into senescence and eventually dies. Figure created by Michael Fossel, MD, PhD.
The beginnings of telomere-targeted therapies
The number of publications on telomerase and its different
biological effects has immensely increased since the dawn
of the 2000s [99]. Since the discovery of telomerase, its
impact diverged into two schools of thought. The first
school is “pro-TERT,” favoring its regenerative function
at shortened telomeres, extending the cell’s population
doubling, and restoring mitotic capacity. The other school
is mainly “anti-TERT”, which extends to applying knowledge of telomeres, telomerase, and their investment in the
cell’s homeostatic profile in treating miscellaneous cancerous diseases. In fact, the anti-telomerase therapies have
been widely exploited in cancer research in the last three
decades.
Anti-telomerase molecules have been chosen by selected
biotechnology companies as potential drugs, mostly
targeted against cancerous tissues [100-102]. One such
pharmaceutical, N3’-P5’ thio-phosphoramidate (NPS) oligonucleotide (GRN163) acting as a telomerase antagonist,
although primarily shown as a competitive and efficient
cancer drug introduced by Geron Corporation, had turned
out to deliver no expected results. Interestingly, the antitelomerase therapies seem to impair the findings of the
opposite school, i.e. upregulating telomerase levels in the
cell for therapeutic reasoning. This only showcases that
the situation is far more complicated than the initial fndings had indicated.
It is still inconclusive and more research must be done to
sincerely state that telomerase itself does not increase the
risk of cancer development, but there are more than plain
premises helping to conclude the germinal fndings about
telomerase’s role in cancer [103]. It must be highlighted
again that it is the optimal telomere length that must be
present in a cancerous cell in order to satisfy the unstoppable growth. If the telomeres were too long, they would
provide the cell with incontestable genomic stability, thus
precluding cancer development [104]. On the other hand,
if the average length of the cancerous cell’s telomeres was
too small, the cell would fall into the senescence route and
eventually die through apoptosis [105, 106]. Therefore,
we should expect different clinical results depending on
the dosage of anti-telomerase therapy, duration, and the
activity of anti-telomerase agents. Perhaps, this sort of
cancer treatment would yield positive clinical effects in
a short run, but the long-term consequences seen in the
patient are difficult to predict, especially if to consider
the complexity of the tumorigenesis process and the still
uncertain role of telomeres and telomerase in its development.
Thereby, instead of focusing sole attention on cancer in
the light of telomerase, we should consider the telomerase
therapeutic methods that could potentially help confine
age-related and other degenerative diseases. Some agree
that telomere extension through the reverse transcription
of telomerase complex outweighs the risk of cancer promotion [107].
Transient transfection of somatic cells with telomerase transcript, full or truncated, has shown that 1) a causation
relationship exists between the length of telomeres and
the expressed levels of telomerase, and 2) the extrinsic
delivery of telomerase to the cells restores their function,
but does not change their phenotypes [26, 108, 109]. The
frst proof was presented by Bodnar’s laboratory in 1998,
when human retinal pigment cells and foreskin fbroblasts
were transfected with the hTERT-SV40 construct, and
established the laboratory immortal cell lines [11]. Bodnar’s work initiated a cascade of future experiments using
extrinsic telomerase catalytic subunit as the cells’ ‘transformer’, establishing immortalized and phenotypically
regular transformed cells [110-112]. The groundbreaking
experiment presented by DePinho’s lab using a special
mural TERT transcript engineered with a knock-in TERTestrogen receptor (mTERT-ER) allele controlled by an extrinsic estrogen receptor modulator, 4-hydroxytamoxifen
(4-OHT) showed how reversing the systemic degenerative
phenotypes and restoring telomere length in mice with
various telomere dysfunctions can be achieved by external, dosage-dependent telomerase expression regulation
[92]. Other studies with somatic cells successfully transfected with TERT subunit are reviewed in [107].
From time to time, different therapeutic variations concerning telomerase are reinforced by other biomolecules.
In one such study, telomerase therapy was supported by
ectopic Bcl-2 transfection [113]. Telomerase as a biomolecule in tissue engineering and regenerative medicine
(TERM) has already been strongly suggested [114, 115].
Moreover, it is commonly agreed that transient, ectopic
delivery of telomerase under constant physiological control is the only acceptable way for a therapeutic approach.
This model, aided by viral vector technology, had been
well-tested in the past [50, 116, 117]. The same model, but
modifed and extended, works perfectly fne with adenoassociated virus (AAV) as a viral vector for ectopic TERT
delivery, which does not cause tumor transformation or
any genomic instabilities within the transfected cells [93].
Those advancements show that we are ready to embrace
the experimental data coming from the telomere research
and apply this knowledge to clinics, where the patients
could beneft from the billions of dollars already invested
in the telomerase, cancer, and gene therapy research.
The future of ageing: Telomerase therapy aims
Telomerase therapy is rooted in the molecular foundations
of cellular pathology. As such, it might target multiple
potential diseases directly associated with genomic instability and/or TLM disruption. Every age-related disorder
with an etiology correlated with telomere shortening,
supported by experimental and clinical data, can become
a potential target for therapy. The therapy may not only
be used to cure age-related diseases, but it also can be applied to prevent their occurrence. The premise is clear: if
we can suppress genomic instability and strengthen cellular homeostasis, the risks of disease onset and its progression might be reduced.
Previously in the text, reversing cellular senescence was
mentioned. This is an important aspect of every biogerontological therapy which must be addressed with high prudence. From a biomedical standpoint, we are currently not
able to reverse molecular ageing in terms of the simple
understanding: one cannot reverse the biological clock as
this would stand against the second law of thermodynamics. However, one could help suppress the somatic damage to the genome and prevent further deleterious changes
happening at the molecular level. These actions will reverse the molecular hallmarks of cellular senescence, giving an individual a better quality of living.
Moreover, as it was already discussed, a strong correlation between cancerous transformation and the telomerase apparatus control exists. If the total loss of telomeric
repeats in a given set of chromosomes dictates further
mechanisms being activated and deactivated during rigorous checkpoints of the cell cycle, preventing the cell from
telomere truncation would yield broadly positive clinical
outcomes for cancer. More experimental data is needed to
draw proper conclusions, and it is still too early to declare
any fnal statement on the correlation between cancer development and telomere length control, but based on the
data we have at the moment we can conclude that there
is a promising platform for telomerase therapy targeting
cancerous changes at the molecular level. In such terms,
telomerase-based gene therapy could become a splendid
showcase of the potential of telomerase: to cure age-related disorders and to protect from cancer using one single
therapeutic platform.
Evaluatng telomerase therapy
Human clinical studies must be completed before we can
state that the therapy is successful and rational. Predictions set a new course of developing gene therapy for
age-related diseases, where a biopsychosocial model of
disorder must be evaluated for the full understanding of
potential telomerase therapy. Telocyte, an American biotechnology company, is one of the frst commercial entities focused on producing an effective telomerase-based
gene therapy platform to cure Alzheimer’s disease and
other age-related diseases. As discussed thoroughly by
Fossel in his recent publication on the role of telomeres
in developing Alzheimer’s disease and other dementias,
even the ARDs of the brain, with the main functional
cells, neurons, mitotically inactive and fully differentiated,
can still be explained on the basis of gradually shortening
telomeres by taking into account the remaining 80% of
the brain’s glial cells which are capable of mitotic division
[52]. Because glial cells are supportive cells, their morphological and physiological deterioration affects the neural cells more than it had been predicted [118, 119]. If the
main upstream cause of age-associated dementias are the
senescing glial (supportive) cells, whose deteriorated state
stems from genomic instability triggered mainly by short ening telomeres, in this case the progressive functional
failure of neurons and the occurrence of the hallmarks of
Alzheimer’s disease (tau tangles and β-amyloid plaques)
can be fully understood, pathophysiologically, through
the biology of telomeres. Therefore, Telocyte’s prototypic
treatment system is based on extracellular telomerase transcript delivery. It will be done through the AAV-9 vector
injected into the spinal fluid and acting transiently on the
glial cells. If successful, this gene therapy approach builds
a bigger therapeutic platform which will serve to eventually cure most age-related disorders.
The biology of ageing is a crossroad for miscellaneous
felds of medicine and biology, where one must not speak
about one cause, one effect or a simplified correlational
model. Through the use of the powerful proteomics isolated chromatin segments (PICh) method, it has been found
that three cell lines manifesting two types of the TLM:
telomerase expression and the ALT, were associated with
~400 different proteins, sharing 98 proteins in common
[118]. Many of those proteins could be associated with
different genomic loci, cell cycle stages and chromatin
looping effects, but this approximation showcases the
great abundance of regulatory elements involved directly
or indirectly in the processes of telomere length regulation
(TRF1, POT1, Apollo), DNA damage mechanisms (Rad50,
BLM, ERCC1, PARP1), chromosome end protection (Ku
70, Ku 80, ATM, ATR, WRN), and many others [119-121].
Large numbers of proteins on the cross-talk of different
paths must be counted as well, which only adds on to the
complexity of the biology of telomeres.
Conclusions
Should a therapy based solely on telomerase be trusted?
This question requires a defnitive answer based only on
data; no speculations have room here. However, we are
certain that telomerase therapy might become the gold
standard for all age-related diseases in the next 10 years.
Recent findings (cited elsewhere in the text) undoubtedly showcase the positive role of telomerase in the “old”
and the senescent cells, which retrieve their biological
functions and resupply the pool of “young” and healthy
cells of the tissue. The exact role of telomeres and their
length control in cancer development is still debatable, but
further steps in clinical research will finally resolve this
dilemma.
There is yet much to say in ageing research. New discoveries consistently broaden our understanding of telomere
biology and demonstrate its complexity, which is understandable in terms of its highly signifcant role in homeostasis. It must be noted that, even though many signaling
pathways and metabolic processes are involved, the main
role of telomeres is to protect the chromosomal ends
from degradation and the NHEJ, thus protecting the cell
from genomic instability and physiological deferment. If
this these changes could be stopped and reversed, and the
metabolic environment of the cell stabilized, we should do it. We must push the progress of our own fndings and
apply them in the real world, move outside the laboratory
settings right into the clinics, where the patients are the
fnal recipients of what once was started in laboratory.
Declarations
Authors’ contributions
Steve Liebich is the sole author of the article.
Acknowledgements
Dr. Woodring Wright, a phenomenal aging and cancer researcher who passed away in August, 2019: his inspiration will live on. Dr Michael Fossel, a technical and graphical supporter of this article. The author’s brother whose love will stand the test of time.
Conflict of Interest
The author declares no conflict of interest.
References
1. Blackburn E H, Gall J G. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. Journal of molecular biology, 1978, 120(1): 33-53.
2. Olovnikov A M. A theory of marginotomy: the incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. Journal of theoretical biology, 1973, 41(1): 181-190.
3. Meyne J, Ratliff R L, MoYzIs R K. Conservation of the human telomere sequence (TTAGGG) n among vertebrates. Proceedings of the National Academy of Sciences, 1989, 86(18): 7049-7053.
4. Chong L, Van Steensel B, Broccoli D, et al. A human telomeric protein. Science, 1995, 270(5242): 1663-1667.
5. De Lange, T. (2005). Shelterin: the protein complex that shapes and safeguards human telomeres. Genes & development, 19(18), 2100-2110.
6. Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990; 345: 458.
7. Lindsey J, McGill N I, Lindsey L A, et al. In vivo loss of telomeric repeats with age in humans. Mutation Research/DNAging, 1991, 256(1): 45-48.
8. Allsopp R C, Vaziri H, Patterson C, et al. Telomere length predicts replicative capacity of human fibroblasts. Proceedings of the National Academy of Sciences, 1992, 89(21): 10114-10118.
9. Lombard D B, Chua K F, Mostoslavsky R, et al. DNA repair, genome stability, and aging. Cell, 2005, 120(4): 497- 512.
10. Greider C W, Blackburn E H. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature, 1989, 337(6205): 331-337.
11. Bodnar A G, Ouellette M, Frolkis M, et al. Extension of life-span by introduction of telomerase into normal human cells. science, 1998, 279(5349): 349-352.
12. Cong Y S, Wright W E, Shay J W. Human telomerase and its regulation. Microbiology and molecular biology reviews, 2002, 66(3): 407-425.
13. Collins K, Mitchell J R. Telomerase in the human organism. Oncogene, 2002, 21(4): 564-579.
14. Shay J W, Wright W E. Telomeres and telomerase in normal and cancer stem cells. FEBS letters, 2010, 584(17): 3819-3825.
15. Hayflick L, Moorhead P S. The serial cultivation of human diploid cell strains. Experimental cell research, 1961, 25(3): 585-621.
16. Medvedev Z A. An attempt at a rational classification of theories of ageing. Biological Reviews, 1990, 65(3): 375- 398.
17. Jin K. Modern biological theories of aging. Aging and disease, 2010, 1(2): 72.
18. Harraan D. Aging: a theory based on free radical and radiation chemistry. 1955.
19. Berlett B S, Stadtman E R. Protein oxidation in aging, disease, and oxidative stress[J]. Journal of Biological Chemistry, 1997, 272(33): 20313-20316.
20. Cadenas E, Davies K J A. Mitochondrial free radical generation, oxidative stress, and aging. Free radical biology and medicine, 2000, 29(3-4): 222-230.
21. Pham-Huy L A, He H, Pham-Huy C. Free radicals, antioxidants in disease and health. International journal of biomedical science: IJBS, 2008, 4(2): 89.
22. Fossel M. The Telomerase Revolution: The Enzyme That Holds the Key to Human Aging and Will Soon Lead to Longer, Healthier Lives. BenBella Books, Inc., 2015.
23. Blasco M A. Telomere length, stem cells and aging. Nature chemical biology, 2007, 3(10): 640-649.
24. Blackburn E H, Epel E S, Lin J. Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science, 2015, 350(6265): 1193- 1198.
25. Shay J W, Wright W E. Hallmarks of telomeres in ageing research. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland, 2007, 211(2): 114-123.
26. Morales C P, Holt S E, Ouellette M, et al. Absence of cancer–associated changes in human fibroblasts immortalized with telomerase. Nature genetics, 1999, 21(1): 115- 118.
27. Faragher R G A, Mulholland B, Tuft S J, et al. Aging and the cornea. British journal of ophthalmology, 1997, 81(10): 814-817.
28. Aikata H, Takaishi H, Kawakami Y, et al. Telomere reduction in human liver tissues with age and chronic inflammation. Experimental cell research, 2000, 256(2): 578- 582.
29. Fossel M. Cell senescence in human aging: A review of the theory. In Vivo, 2000, 14(1): 29-34.
30. Fossel M. Use of telomere length as a biomarker for aging and age-related disease. Current Translational Geriatrics and Experimental Gerontology Reports, 2012, 1(2): 121-127.
31. Laberthonnière C, Magdinier F, Robin J D. Bring it to an end: does telomeres size matter? Cells, 2019, 8(1): 30.
32. Calvert P A, Liew T V, Gorenne I, et al. Leukocyte telomere length is associated with high-risk plaques on virtual histology intravascular ultrasound and increased proinflammatory activity. Arteriosclerosis, thrombosis, and vascular biology, 2011, 31(9): 2157-2164.
33. Valdes A M, Andrew T, Gardner J P, et al. Obesity, cigarette smoking, and telomere length in women. The lancet, 2005, 366(9486): 662-664.
34. Shen J, Gammon M D, Terry M B, et al. Telomere length, oxidative damage, antioxidants and breast cancer risk. International journal of cancer, 2009, 124(7): 1637- 1643.
35. Starr J M, Shiels P G, Harris S E, et al. Oxidative stress, telomere length and biomarkers of physical aging in a cohort aged 79 years from the 1932 Scottish Mental Survey. Mechanisms of ageing and development, 2008, 129(12): 745-751.
36. Ilmonen P, Kotrschal A, Penn D J. Telomere attrition due to infection. PloS one, 2008, 3(5): e2143.
37. Plunkett F J, Franzese O, Belaramani L L, et al. The impact of telomere erosion on memory CD8+ T cells in patients with X-linked lymphoproliferative syndrome. Mechanisms of ageing and development, 2005, 126(8): 855-865.
38. Effros R B, Allsopp R, Chiu C, et al. Shortened telomeres in the expanded CD28-CD8+ cell subset in HIV disease implicate replicative senescence in HIV pathogenesis. Aids, 1996, 10(8): F17-22.
39. Aviv A, Chen W, Gardner J P, et al. Leukocyte telomere dynamics: longitudinal findings among young adults in the Bogalusa Heart Study. American journal of epidemiology, 2009, 169(3): 323-329.
40. Matsubara Y, Murata M, Yoshida T, et al. Telomere length of normal leukocytes is affected by a functional polymorphism of hTERT. Biochemical and biophysical research communications, 2006, 341(1): 128-131.
41. Nordfjäll K, Osterman P, Melander O, et al. hTERT− 1327T/C polymorphism is not associated with age-related telomere attrition in peripheral blood. Biochemical and biophysical research communications, 2007, 358(1): 215-218.
42. Zhang D, Wen X, Zhang L, et al. DNA methylation of human telomerase reverse transcriptase associated with leukocyte telomere length shortening in hyperhomocysteinemia-type hypertension in humans and in a rat model. Circulation Journal, 2014: CJ-14-0233.
43. Brouilette S W, Moore J S, McMahon A D, et al. Telomere length, risk of coronary heart disease, and statin treatment in the West of Scotland Primary Prevention Study: a nested case-control study. The Lancet, 2007, 369(9556): 107-114.
44. Valdes A M, Richards J B, Gardner J P, et al. Telomere length in leukocytes correlates with bone mineral density and is shorter in women with osteoporosis. Osteoporosis International, 2007, 18(9): 1203-1210.
45. Sampson M J, Winterbone M S, Hughes J C, et al. Monocyte telomere shortening and oxidative DNA damage in type 2 diabetes. Diabetes care, 2006, 29(2): 283-289.
46. Fitzpatrick A L, Kronmal R A, Kimura M, et al. Leukocyte telomere length and mortality in the Cardiovascular Health Study. Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences, 2011, 66(4): 421-429.
47. Savale L, Chaouat A, Bastuji-Garin S, et al. Shortened telomeres in circulating leukocytes of patients with chronic obstructive pulmonary disease. American journal of respiratory and critical care medicine, 2009, 179(7): 566- 571.
48. Cronkhite J T, Xing C, Raghu G, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. American journal of respiratory and critical care medicine, 2008, 178(7): 729-737.
49. Scheinberg P, Cooper J N, Sloand E M, et al. Association of telomere length of peripheral blood leukocytes with hematopoietic relapse, malignant transformation, and survival in severe aplastic anemia. Jama, 2010, 304(12): 1358-1364.
50. Funk, W. D., Wang, C. K., Shelton, D. N., Harley, C. B., Pagon, G. D., & Hoeffler, W. K. (2000). Telomerase expression restores dermal integrity to in vitro-aged fibroblasts in a reconstituted skin model. Experimental cell research, 258(2), 270-278.
51. Fyhrquist F, Saijonmaa O, Strandberg T. The roles of senescence and telomere shortening in cardiovascular disease. Nature Reviews Cardiology, 2013, 10(5): 274-283.
52. Fossel M. A unified model of dementias and age-related neurodegeneration. Alzheimer’s & Dementia, 2020, 16(2): 365-383.
53. Fossel M. Telomerase and the aging cell: implications for human health. JAMA, 1998, 279(21): 1732-1735.
54. Vaziri H, Dragowska W, Allsopp R C, et al. Evidence for a mitotic clock in human hematopoietic stem cells: loss of telomeric DNA with age. Proceedings of the National Academy of Sciences, 1994, 91(21): 9857-9860.
55. Zhang J, Li L. Stem cell niche: microenvironment and beyond. Journal of Biological Chemistry, 2008, 283(15): 9499-9503.
56. Sharpless N E, DePinho R A. How stem cells age and why this makes us grow old. Nature reviews Molecular cell biology, 2007, 8(9): 703-713.
57. Cong Y S, Wen J, Bacchetti S. The human telomerase catalytic subunit hTERT: organization of the gene and characterization of the promoter. Human molecular genetics, 1999, 8(1): 137-142.
58. Liebich S. hTERT Promoter Regulation by Differentiation Mechanisms vs Telomerase Activity in Somatic, Embryonic, and Cancerous Cells. OBM geriatrics. 2019; 3(2):1- 14.
59. Wright W E, Piatyszek M A, Rainey W E, et al. Telomerase activity in human germline and embryonic tissues and cells. Developmental genetics, 1996, 18(2): 173-179.
60. Liu K, Schoonmaker M M, Levine B L, et al. Constitutive and regulated expression of telomerase reverse transcriptase (hTERT) in human lymphocytes. Proceedings of the National Academy of Sciences, 1999, 96(9): 5147-5152.
61. Shay J W, Bacchetti S. A survey of telomerase activity in human cancer. European journal of cancer, 1997, 33(5): 787-791.
62. Kim N W, Piatyszek M A, Prowse K R, et al. Specific association of human telomerase activity with immortal cells and cancer. Science, 1994, 266(5193): 2011-2015.
63. Artandi S E, DePinho R A. Telomeres and telomerase in cancer. Carcinogenesis, 2010, 31(1): 9-18.
64. Liebich S. hTERT Promoter Regulation by Differentiation Mechanisms vs Telomerase Activity in Somatic, Embryonic, and Cancerous Cells. OBM Geriatrics 2019;3(2):14.
65. Ulaner G A, Hu J F, Vu T H, et al. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer research, 1998, 58(18): 4168-4172.
66. Wang J, Xie L Y, Allan S, et al. Myc activates telomerase. Genes & development, 1998, 12(12): 1769-1774.
67. Xu D, Popov N, Hou M, et al. Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentiation of HL60 cells. Proceedings of the National Academy of Sciences, 2001, 98(7): 3826-3831.
68. Ramlee M K, Wang J, Toh W X, et al. Transcription regulation of the human telomerase reverse transcriptase (hTERT) gene. Genes, 2016, 7(8): 50.
69. Cong Y S, Wright W E, Shay J W. Human telomerase and its regulation. Microbiology and molecular biology reviews, 2002, 66(3): 407-425.
70. Cairney C J, Keith W N. Telomerase redefined: integrated regulation of hTR and hTERT for telomere maintenance and telomerase activity. Biochimie, 2008, 90(1): 13-23.
71. Palm W, de Lange T. How shelterin protects mammalian telomeres. Annual review of genetics, 2008, 42: 301-334.
72. Khanna K K, Jackson S P. DNA double-strand breaks: signaling, repair and the cancer connection. Nature genetics, 2001, 27(3): 247-254.
73. Abraham R T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes & development, 2001, 15(17): 2177-2196.
74. Jackson S P. Sensing and repairing DNA double-strand breaks. Carcinogenesis, 2002, 23(5): 687-696.
75. Kim W, Ludlow A T, Min J, et al. Regulation of the human telomerase gene TERT by telomere position effect—over long distances (TPE-OLD): implications for aging and cancer. PLoS biology, 2016, 14(12): e2000016.
76. Bouwman B A M, de Laat W. Getting the genome in shape: the formation of loops, domains and compartments. Genome biology, 2015, 16(1): 1-9.
77. Zou Y, Sfeir A, Gryaznov S M, et al. Does a sentinel or a subset of short telomeres determine replicative senescence? Molecular biology of the cell, 2004, 15(8): 3709- 3718.
78. Vera E, de Jesus B B, Foronda M, et al. The rate of increase of short telomeres predicts longevity in mammals. Cell reports, 2012, 2(4): 732-737.
79. Muñoz-Lorente M A, Martinez P, Tejera A, et al. AAV9- mediated telomerase activation does not accelerate tumorigenesis in the context of oncogenic K-Ras-induced lung cancer. PLoS genetics, 2018, 14(8): e1007562.
80. Hanahan D, Weinberg R A. The hallmarks of cancer Cell 100 (1): 57–70. Find this article online, 2000.
81. Perrem K, Bryan T M, Englezou A, et al. Repression of an alternative mechanism for lengthening of telomeres in somatic cell hybrids[J]. Oncogene, 1999, 18(22): 3383- 3390.
82. Cesare A J, Reddel R R. Alternative lengthening of telomeres: models, mechanisms and implications. Nature reviews genetics, 2010, 11(5): 319-330.
83. Rudd M K, Friedman C, Parghi S S, et al. Elevated rates of sister chromatid exchange at chromosome ends. PLoS Genet, 2007, 3(2): e32.
84. Feldser D M, Greider C W. Short telomeres limit tumor progression in vivo by inducing senescence. Cancer cell, 2007, 11(5): 461-469.
85. Blasco M A. Telomeres and human disease: ageing, cancer and beyond. Nature Reviews Genetics, 2005, 6(8): 611-622.
86. Bolzán A D, Bianchi M S. Telomeres, interstitial telomeric repeat sequences, and chromosomal aberrations. Mutation Research/Reviews in Mutation Research, 2006, 612(3): 189-214.
87. Blackburn E H, Szostak J W. The molecular structure of centromeres and telomeres. Annual review of biochemistry, 1984, 53(1): 163-194.
88. Zhan W H, Ma J P, Peng J S, et al. Telomerase activity in gastric cancer and its clinical implications. World journal of gastroenterology, 1999, 5(4): 316.
89. Counter C M, Hirte H W, Bacchetti S, et al. Telomerase activity in human ovarian carcinoma. Proceedings of the National Academy of Sciences, 1994, 91(8): 2900-2904.
90. Martins C S, Santana-Lemos B A, Saggioro F P, et al. Telomere length and telomerase expression in pituitary tumors. Journal of endocrinological investigation, 2015, 38(11): 1243-1246.
91. Scheinberg P, Cooper J N, Sloand E M, et al. Association of telomere length of peripheral blood leukocytes with hematopoietic relapse, malignant transformation, and survival in severe aplastic anemia. Jama, 2010, 304(12): 1358-1364.
92. Jaskelioff M, Muller F L, Paik J H, et al. Telomerase reactivation reverses tissue degeneration in aged telomerasedeficient mice. Nature, 2011, 469(7328): 102-106.
93. Bernardes de Jesus B, Vera E, Schneeberger K, et al. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO molecular medicine, 2012, 4(8): 691-704.
94. Jiang X R, Jimenez G, Chang E, et al. Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nature genetics, 1999, 21(1): 111-114.
95. Counter C M, Avilion A A, LeFeuvre C E, et al. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. The EMBO journal, 1992, 11(5): 1921-1929.
96. Park J I, Venteicher A S, Hong J Y, et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature, 2009, 460(7251): 66-72.
97. Varela E, Munoz-Lorente M A, Tejera A M, et al. Generation of mice with longer and better preserved telomeres in the absence of genetic manipulations. Nature communications, 2016, 7(1): 1-16.
98. Tomás-Loba A, Flores I, Fernández-Marcos P J, et al. Telomerase reverse transcriptase delays aging in cancerresistant mice. Cell, 2008, 135(4): 609-622.
99. Corey D R. Telomeres and telomerase: from discovery to clinical trials. Chemistry & biology, 2009, 16(12): 1219- 1223.
100.Shea-Herbert B, Pongracz K, Shay J W, et al. Oligonucleotide N3′→ P5′ phosphoramidates as efficient telomerase inhibitors. Oncogene, 2002, 21(4): 638-642.
101.Corey D R. Chemical modification: the key to clinical application of RNA interference? The Journal of clinical investigation, 2007, 117(12): 3615-3622.
102.Asai A, Oshima Y, Yamamoto Y, et al. A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer research, 2003, 63(14): 3931-3939.
103.Harley C B. Telomerase is not an oncogene. Oncogene, 2002, 21(4): 494-502.
104.Murnane J P. Telomere dysfunction and chromosome instability. Mutation research/Fundamental and molecular mechanisms of mutagenesis, 2012, 730(1-2): 28-36.
105.Bernadotte A, Mikhelson V M, Spivak I M. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging (Albany NY), 2016, 8(1): 3.
106.Herbig U, Jobling W A, Chen B P C, et al. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21CIP1, but not p16INK4a. Molecular cell, 2004, 14(4): 501-513.
107.Harley C B. Telomerase therapeutics for degenerative diseases. Current Molecular Medicine, 2005, 5(2): 205-211.
108.Poh M, Boyer M, Solan A, et al. Blood vessels engineered from human cells. The Lancet, 2005, 365(9477): 2122- 2124.
109.Klinger R Y, Blum J L, Hearn B, et al. Relevance and safety of telomerase for human tissue engineering. Proceedings of the National Academy of Sciences, 2006, 103(8): 2500-2505.
110.Steinert S, Shay J W, Wright W E. Transient expression of human telomerase extends the life span of normal human fibroblasts. Biochemical and biophysical research communications, 2000, 273(3): 1095-1098.
111.Wyllie F S, Jones C J, Skinner J W, et al. Telomerase prevents the accelerated cell ageing of Werner syndrome fibroblasts. Nature genetics, 2000, 24(1): 16-17.
112.Condon J, Yin S, Mayhew B, et al. Telomerase immortalization of human myometrial cells. Biology of reproduction, 2002, 67(2): 506-514.
113.Petersen T, Niklason L. Cellular lifespan and regenerative medicine. Biomaterials, 2007, 28(26): 3751-3756.
114.Shay J W, Wright W E. Use of telomerase to create bioengineered tissues. Annals of the New York Academy of Sciences, 2005, 1057(1): 479-491.
115.Jäger K, Walter M. Therapeutic targeting of telomerase. Genes, 2016, 7(7): 39.
116.Murasawa S, Llevadot J, Silver M, et al. Constitutive human telomerase reverse transcriptase expression enhances regenerative properties of endothelial progenitor cells. circulation, 2002, 106(9): 1133-1139.
117.Verra N C V, Jorritsma A, Weijer K, et al. Human telomerase reverse transcriptase-transduced human cytotoxic T cells suppress the growth of human melanoma in immunodeficient mice. Cancer Research, 2004, 64(6): 2153- 2161.
118.Boccardi V, Pelini L, Ercolani S, et al. From cellular senescence to Alzheimer’s disease: The role of telomere shortening. Ageing research reviews, 2015, 22: 1-8.
119.Flanary B E, Sammons N W, Nguyen C, et al. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation research, 2007, 10(1): 61-74.
120.Déjardin J, Kingston R E. Purification of proteins associated with specific genomic Loci. Cell, 2009, 136(1): 175- 186.
121.De Boeck G, Forsyth R G, Praet M, et al. Telomere-associated proteins: cross-talk between telomere maintenance and telomere-lengthening mechanisms. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland, 2009, 217(3): 327-344.