Open Access | Model Profile
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
A mouse model of sleep deprived neuropathology to study resilience to Alzheimer’s disease
* Corresponding author: Warren Ladiges
Mailing address: Department of Comparative Medicine, School
of Medicine, University of Washington, Seattle, WA, 98195,
USA.
E-mail: wladiges@uw.edu
Received: 31 May 2020 / Accepted: 06 June 2020
DOI: 10.31491/APT.2020.06.025
Abstract
Resilience to Alzheimer’s disease (AD) is a well-known clinical and pathological observation, but the mechanisms involved are not known. Adequate sleep is a potential factor in maintaining resilience to neurodegenerative conditions such as AD. It is well known that sleep deprivation is a major health concern in developed countries and is associated with increasing age. Normal aging produces sleep disturbances including sleep fragmentation and sleep loss in humans, which has recently been recognized as an important risk factor for AD. The idea of enhancing AD resilience by targeting sleep deprivation encompasses the concept of physical resilience to aging. We demonstrate the detrimental effects of sleep deprivation in aging mice and propose a mouse model of AD to test the concept. The model provides a means of testing therapeutics that could be investigated in clinical trials designed to prevent sleep deprivation and enhance resilience to aging and AD in the elderly.
Keywords
Mouse model of sleep deprivation, resilience to aging, resilience to Alzheimer’s disease
The prevalence of neurodegenerative diseases such as
Alzheimer’s disease (AD) is expected to soar with the
number of elderly individuals in both developed and
developing countries now rising dramatically. Efforts to
find disease-modifying treatments have been largely unsuccessful in part due to inability to assess early signs of
disease and risk factors associated with increasing age,
and the lack of predictable preclinical animal models. One
approach to investigating risk factors for AD is to look at
attributes that oppose risk, ie, resilience. Resilience is the
ability of an organism to successfully respond and recover
from physical stress. The occurrence of resilience to AD
has been suggested based on the absence of clinical signs
of cognitive impairment but presence of neuropathological lesions typical of AD at autopsy. The causes for this
apparent paradox are not known, but resilience to physical
stress could play a role. A good example of a type of stress
that has neurological effects is sleep deprivation.
It is fairly well established that sleep disturbances in crease the risk of dementia and AD and there is growing
evidence that poor sleep leads to acceleration in the progression of neurodegenerative disorders and may play a
role in pathogenesis. Clinical studies are well supported
by animal studies showing that sleep deprivation induces
learning and memory dysfunction and exacerbates ADlike pathologies in AD transgenic mice [1]. Therefore,
sleep deprivation is a physical stressor that decreases resilience to healthy aging and increases the risk for AD (Figure
1). Prevention of the adverse effects of sleep deprivation
would be a logical approach to enhance resilience to AD
by enhancing resilience to aging.
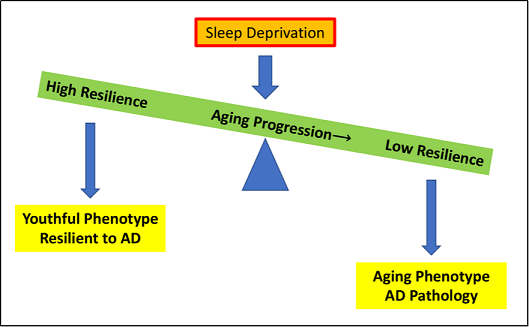
Figure 1. As aging progresses, the physical resilience of an individual towards aging and sleep deprivation decreases, tipping the scale and leading to an aging phenotype and the development of AD pathology. Some individuals continue to maintain or regain functions quickly following insults as age increases, producing a more youthful phenotype and resilience to AD.
Preclinical models for investigating resilience to aging and
AD are not well described. We have developed an aging
mouse model of short-term sleep deprivation that results
in neurodegenerative changes and cognitive impairment
[2]. We suspected that sleep deprivation would adversely
impact synaptic function through mitochondrial disruption. Mitochondrial dysfunction leading to decreased ATP
production and increased ROS resulting from impaired
electron transport chain function appears prominently in
both aging and AD [3-5]. We showed that sleep-deprived
mice had signifcantly higher levels of mitochondrial ROS
production and a significant decrease in ATP synthesis
in the brain compared with non-sleep deprived mice [6].
Closely linked with mitochondrial dysfunction, our mouse
model of short-term sleep deprivation showed that learning impairment was associated with mechanisms related to synaptic plasticity in the hippocampus. N-methyl-D-aspartate receptor, a well-known synaptic glutamate receptor
that regulates long-term potentiation and synaptic plasticity [7-8], and brain derived neurotropic factor, a supportive regulator of synaptic plasticity, were both signifcantly
decreased. In addition, neuroinflammatory cytokine levels
of MCP-1, TNF-ɑ, and IL-6 were increased.
This aging mouse model of short-term sleep deprivation
provides an excellent background for studying effects on
pathogenesis of AD. Certainly it would be applicable to
currently available transgenic AD mouse models. However, in most of the transgenic lines, a signifcant increase
in APP production begins early in life possibly in utero,
which may trigger consequences that alter aging and the
rate of aging, and may not mimic the biochemical changes
observed in AD. Most importantly, it is problematic to
measure early events in the development of AD with
increasing age in these models. Desirable features of a
model system would allow for a precisely controlled challenge time and onset of disease in an aging background.
We have shown that introduction of Aβ42/P301Ltau into
the brains of older mice results in cognitive impairment
and neuropathology including inflammation, neuronal
degeneration, synaptic dysfunction, and vascular impairment (unpublished data). Effective use of this model does
require access to aging mice, AAV vectors, and expertise
in stereotactic injections into specifc regions of the mouse
brain [9]. Whatever the AD model, short-term sleep deprivation provides a highly informative and rapid model to
investigate ways of enhancing resilience to aging and AD
by preventing sleep deprived neuropathology with drugs
or other intervention measures (Figure 2).
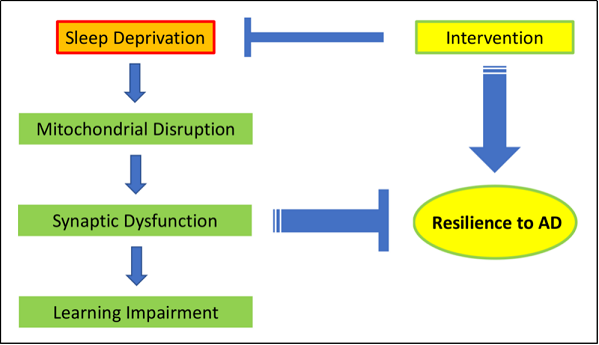
Figure 2. Short term sleep deprivation induces a cascade of molecular events starting with mitochondrial disruption to synaptic dysfunction and subsequent learning impairment. The neurological effects of sleep deprivation could intensify over time and decrease resilience to AD. Intervention strategies that prevent neuronal pathology induced by sleep deprivation in aging mice would have the potential to alleviate cognitive impairment and increase resilience to AD.
Declaration
Conflict of Interest
The authors declare that they have no conflict of interest.
References
1. Di Meco A, Joshi Y B, Praticò D. Sleep deprivation impairs memory, tau metabolism, and synaptic integrity of a mouse model of Alzheimer’s disease with plaques and tangles. Neurobiology of aging, 2014, 35(8): 1813-1820.
2. Mukherjee K, Lee A, Zhu L, et al. Sleep-deprived cognitive impairment in aging mice is alleviated by rapamycin. Aging Pathobiology and Therapeutics, 2019, 1(1): 05-09.
3. Salminen A, Haapasalo A, Kauppinen A, et al. Impaired mitochondrial energy metabolism in Alzheimer’s disease: Impact on pathogenesis via disturbed epigenetic regulation of chromatin landscape. Progress in neurobiology, 2015, 131: 1-20.
4. Stahon K E, Bastian C, Griffith S, et al. Age-related changes in axonal and mitochondrial ultrastructure and function in white matter. Journal of Neuroscience, 2016, 36(39): 9990-10001.
5. Pérez M J, Ponce D P, Osorio-Fuentealba C, et al. Mitochondrial bioenergetics is altered in fibroblasts from patients with sporadic Alzheimer’s disease. Frontiers in neuroscience, 2017, 11: 553.
6. Wu J, Dou Y, Ladiges W C. Adverse neurological effects of short-term sleep deprivation in aging mice are prevented by SS31 peptide. bioRxiv, 2020.
7. Lüscher C, Malenka R C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/ LTD). Cold Spring Harbor perspectives in biology, 2012, 4(6): a005710.
8. Silva A J. Molecular and cellular cognitive studies of the role of synaptic plasticity in memory. Journal of neurobiology, 2003, 54(1): 224-237.
9. Darvas M, Keene D, Ladiges W. A geroscience mouse model for Alzheimer’s disease. Pathobiology of Aging and Age-Related Diseases, 2019.