Open Access | Review
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
Thioredoxin and aging: What have we learned from the survival studies?
* Corresponding author: Yuji Ikeno, M.D., Ph.D.
Mailing address: Barshop Institute for Longevity and Aging
Studies, The University of Texas Health Science Center at San
Antonio, 15355 Lambda Drive, San Antonio, TX 78245-3207,
USA.
E-mail: ikeno@uthscsa.edu
Received: 03 June 2020 / Accepted: 11 September 2020
DOI: 10.31491/APT.2020.09.028
Abstract
Our laboratory has conducted the first systematic survival studies to examine the biological effects of the antioxidant protein thioredoxin (Trx) on aging and age-related pathology. Our studies with C57BL/6 mice overexpressing Trx1 [Tg(act-TRX1)+/0 and Tg(TXN)+/0] demonstrated a slight extension in early lifespan compared to wild-type (WT) mice; however, no significant effects were observed in the later part of life. Overexpression of Trx2 in male C57BL/6 mice [Tg(TXN2)+/0] demonstrated a slightly extended lifespan compared to WT mice. The pathology results from two lines of Trx1 transgenic mice showed a slightly higher incidence of age-related neoplastic diseases compared to WT mice, and a slight increase in the severity of lymphoma, a major neoplastic disease, was observed in Trx2 transgenic mice. Together these studies indicate that Trx overexpression in one compartment of the cell (cytosol or mitochondria alone) has marginal beneficial effects on lifespan. On the other hand, down-regulation of Trx in either the cytosol (Trx1KO) or mitochondria (Trx2KO) showed no significant changes in lifespan compared to WT mice, despite several changes in pathophysiology of these knockout mice. When we examined the synergetic effects of overexpressing Trx1 and Trx2, TXNTg x TXN2Tg mice showed a significantly shorter lifespan with accelerated cancer development compared to WT mice. These results suggest that synergetic effects of Trx overexpression in both the cytosol and mitochondria on aging are deleterious and the development of age-related cancer is accelerated. On the other hand, we have recently found that down-regulation of Trx in both the cytosol and mitochondria in Trx1KO x Trx2KO mice has beneficial effects on aging. The results generated from our lab along with our ongoing study using Trx1KO x Trx2KO mice could elucidate the key pathways (i.e., apoptosis and autophagy) that prevent accumulation of damaged cells and genomic instability leading to reduced cancer formation.
Keywords
Thioredoxin, transgenic mouse, knockout mouse, cancer, lifespan, aging
Thioredoxin and aging
Initially discovered in the early 1960s, Trx is a small protein (12kDa) that acts as a reductant for a variety of
enzymes [1-7]. It contains two redox-active cysteine residues in the active center (Cys-Gly-Pro-Cys). In humans,
two forms of protein have been identifed: one in cytosol
(Trx1) [8] and another in mitochondria (Trx2) [9]. Trx
is important in maintaining a reduced intracellular environment, which is accomplished through thiol-disulfide
exchange reactions [1]. This unique feature allows Trx to
efficiently control protein function by altering the redox
state of structural or catalytic SH groups. Therefore, Trx
may have an important function in development of agerelated pathophysiological changes through attenuation of oxidative stress/damage or alteration of redox state in
various signaling pathways.
Previous studies conducted in Caenorhabditis elegans
and Drosophila melanogaster have shown that Trx plays
important roles in oxidative stress/damage and longevity
[10, 11]. However, the exact roles of Trx in aging and agerelated diseases in mammals and its effect on crucial signaling pathways have not been fully explored. Therefore,
our laboratory has conducted the frst systematic study to
examine the effects of Trx on survival using mice overexpressing or down-regulating Trx1 (cytosol) and Trx2
(mitochondria).
Here, we review the survival studies conducted in our
laboratory and discuss what we have learned about the effects of Trx on aging and age-related changes.
Transgenic mice/rats overexpressing thioredoxin 1 (cytosol)
The frst survival study to test the effects of Trx1 overexpression was conducted using a transgenic mouse model
generated with human TRX1 cDNA and the β-actin promoter [Tg(act-TRX1)+/0] [12]. An extension in lifespan
and increased resistance to oxidative stress were observed
in Tg(act-TRX1)+/0 mice compared to WT mice [13,14].
Although these observations were very exciting, it is
worth noting that the lifespan of WT mice (C57BL/6) in
this study (approximately 23 months) was considerably
shorter than expected from other studies using C57BL/6
mice housed in barrier facilities, which suggests that conditions for the survival experiment were not optimal. The
median lifespan of C57BL/6 mice in our aging colonies,
for example, is approximately 29–30 months. Thus, it was
necessary to conduct an additional survival study using
Tg(act-TRX1)+/0 mice to determine the effects of overexpressing Trx1 under defned pathogen-free housing conditions.
Our study showed that Trx1 overexpression in Tg(actTRX1)+/0 mice resulted in a higher resistance to oxidative
stress and lower levels of oxidative damage to proteins
and lipids [15]. However, our survival study examining male Tg(act-TRX1)+/0 mice indicated a significant
life-extension only during the first half of their lifespan
compared to WT mice, and no significant changes were
observed thereafter. To further support these initial observations, we conducted another survival study with both
male and female mice [15].
To determine why beneficial effects of Trx1 overexpression were observed only early in life, we examined
whether: 1) the levels of Trx1 changed during aging; and
2) Trx1 overexpression altered age-related pathology.
Our data demonstrated that Tg(act-TRX1)+/0 mice showed
significant age-related decline of Trx1 overexpression
as well as less reduction in protein oxidation levels in
the later part of life [15]. This result is likely explained
by the use of the β-actin promoter to control decreased
expression of the transgene with age. End-of-life pathology data for these mice showed that: 1) the incidence of
lung inflammation was significantly reduced in young
Tg(act-TRX1)+/0 mice; and 2) the incidence of total fatal tumors and lymphomas were slightly higher in old
Tg(act-TRX1)+/0 mice compared to WT littermates [15].
Therefore, Tg(act-TRX1)+/0 mice signifcantly increased in
survival only during the frst half of their lifespan because
of an age-related reduction of Trx1 overexpression and/or
enhanced tumor formation in old mice [15].
Next, we generated a new line of Trx1 transgenic mice
(Tg(TXN)+/0) in order to determine whether continuous overexpression of Trx1 throughout life could extend
maximum lifespan. These mice were generated using a
fragment of the human genome containing the TXN gene
[16]. Tg(TXN)+/0 mice showed significantly higher (approximately 20% to 40%) levels of Trx1 in the tissues
than WT mice. Trx1 overexpression in Tg(TXN)+/0 mice
was maintained up to at least 28-30 months-of-age. The
study showed that the survival curve of Tg(TXN)+/0 mice
was not significantly different from WT mice. Although
the early part of lifespan (75% survival) showed a 6.3%
extension compared to WT mice, no significant life-extending effect was observed over the lifespan [16]. Therefore, our survival data obtained from Tg(TXN)+/0 mice
indicate that continuous Trx1 overexpression in mice had
some benefts only in the early part of life. This observation is similar to the effect of Trx1 overexpression in the
Tg(act-TRX1)+/0 mice.
As mentioned above, the incidence of lung inflammation
in young Tg(act-TRX1)+/0 mice was signifcantly less than
WT mice, and the incidence of total fatal tumors and lymphomas in old Tg(act-TRX1)+/0 mice was slightly higher
than WT mice [15]. These observations are consistent with
some of the biological effects of Trx1, i.e., anti-inflammatory and anti-apoptotic effects. Our previous study showed
lower IL-1β mRNA levels in the liver of Tg(act-TRX1)+/0
mice than WT littermates [15], which could be a contributing factor in the reduction of lung inflammation in Tg(actTRX1)+/0 mice. Additionally, increased levels of the ASK1/
Trx1 complex were observed in Tg(act-TRX1)+/0 mice [15],
which causes inhibition of the apoptosis signal-regulating
kinase-1 (ASK1) pathway [17, 18]. Therefore, Trx1 overexpression in young mice could be beneficial because of
resistance to environmental stresses, including oxidative
stress, and reduced inflammatory changes in the tissues,
including lung. On the other hand, Trx1 overexpression in
old mice may be deleterious because of its anti-apoptotic
action by ASK1 inhibition and promoting cell proliferation, both of which could promote the growth of various
cancers, including lymphoma [8, 19].
In addition to the studies with mice overexpressing Trx1,
we also conducted a survival study with Trx1 transgenic
rats, which were generated using a fragment of the human
genome containing the TXN gene as described above.
We generated transgenic rats using F344 rats because
they are commonly used for aging studies and could allow us to examine the possible species differences of the
role of Trx1 in aging. Our data showed that the levels of Trx1 were significantly higher [approximately 20% to
40%: similar to Tg(TXN)+/0 mice] in all tissues examined
in the Tg(TXN)+/0 rats compared to their WT littermates,
and overexpression of Trx1 was maintained up to 28–30
months of age. Although Tg(TXN)+/0 rats showed similar
Trx1 overexpression to Tg(TXN)+/0 mice, the survival
curve of Tg(TXN)+/0 rats was similar to WT rats, i.e., Trx1
overexpression had little benefcial effects on longevity in
F344 rats (Figure 1).
Overall, the survival studies described above, which were
conducted with two lines of Trx1 transgenic mice, indicate that Trx1 overexpression is benefcial in young mice
and has potential deleterious effects in old mice, possibly
due to the development of different pathophysiological
conditions. Similarly, overexpression of Trx1 in a line of
transgenic rats had little effect on lifespan.
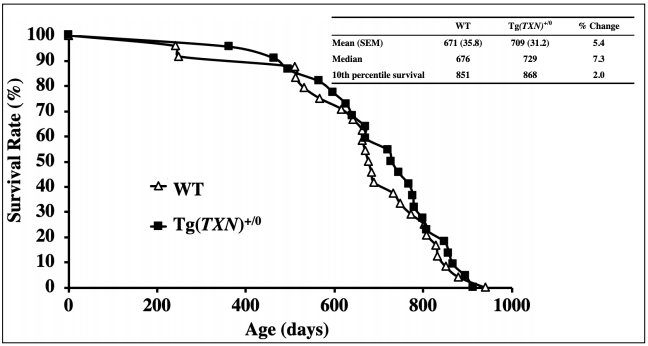
Figure 1. Survival curves of Tg(TXN)+/0 and WT rats. The results from a survival study with male Tg(TXN)+/0 (closed squares) and WT(open triangles) rats are presented. The survival study was conducted with 22 Tg(TXN)v+/0 and 24 WT male rats. No significant difference between Tg(TXN)+/0 and WT rats was observed.
Trx2 transgenic mice overexpressing thioredoxin in mitochondria
The results of the aging study with Trx1 transgenic mice
suggest another possibility, in which Trx overexpression
in mitochondria could be more important in aging. Indeed,
there are several lines of evidence that strongly suggest
that maintaining mitochondrial functions could provide
benefits on age-related pathophysiological changes. For
example, a study with mice overexpressing catalase in
mitochondria (mCAT) showed significant extension of lifespan and attenuated development of some cancers, despite catalase overexpression in the nucleus or peroxisome
showing no changes in lifespan [20]. To further support
the importance of maintaining mitochondrial functions on
pathophysiology, studies have shown that mitochondrial
Trx overexpression: 1) attenuated vascular dysfunction
and hypertension development [21], and 2) maintained
endothelial function and protected against atherosclerosis
development [22]. Thus, we conducted a survival study to test whether mitochondrial Trx overexpression could show
benefcial effects on aging and/or age-related diseases using Trx2 transgenic mice.
Tg(TXN2)+/0 mice were generated with the human thioredoxin 2 (TXN2) gene [23]. The young Tg(TXN2)+/0
mice showed signifcantly higher levels of Trx2 in tissues
examined than WT mice, which was maintained up to
22–24 months old [23]. Tg(TXN2)+/0 mice showed less
hydrogen peroxide production from isolated mitochondria than WT control mice. Despite the reduced hydrogen
peroxide production from mitochondria by Trx2 overexpression, the effects on oxidative damage in the tissues
were disappointing: 1) Tg(TXN2)+/0 mice showed slightly
less (approximately 13–14%) lipid peroxidation measured
by plasma isoprostane levels than WT littermates, and
2) levels of 8-oxodG, a marker of DNA oxidation, were
not changed by Trx2 overexpression. The effects of Trx2
overexpression on signaling pathways were also minimal: 1) mTOR and NFκB pathways were not changed in
Tg(TXN2)+/0 mice compared to WT littermates; 2) Trx2 overexpression increased levels of c-Jun and c-Fos compared to WT mice [23].
Our survival study showed that male Tg(TXN2)+/0 mice
had approximately 8-9% extension of lifespans (the mean,
median, and 10th percentile) compared to WT control
mice. However, these differences were not statistically signifcant [23]. The cross-sectional pathology results showed
that male Tg(TXN2)+/0 mice had a similar number of total
tumors (tumor burden) and incidence of lymphoma compared to WT mice. Although the incidence of lymphoma
was not changed by Trx2 overexpression, Tg(TXN2)+/0
mice showed a slightly higher severity of lymphoma than
WT mice [23]. These pathological analyses suggest that
overexpression of Trx2 in mitochondria may accelerate
age-related lymphoma development, which is similar to
the effects of Trx1 overexpression on tumor development
[15, 16]. As mentioned above, Tg(TXN2)+/0 mice showed
higher levels of c-Jun and c-Fos than WT littermates. Activator protein 1 (AP-1), a complex of proteins of the Jun
and Fos families, binds to TPA-response elements (TRE)
or AP-1 binding sites to transcriptionally activate effector
genes; subsequently, stimulation of cell proliferation and
transformation occur. Thus, increased levels of c-Jun and
c-Fos may be one of the mechanisms that accelerates agerelated tumor development in Tg(TXN2)+/0 mice.
Effects of Trx down-regulation in the cytosol (Trx1) or mitochondria (Trx2) on aging
We also conducted the survival studies to test the effects of thioredoxin down-regulation in the cytosol (Trx1) or
mitochondria (Trx2) on aging. Because Trx1 or Trx2 null
mice are embryonically lethal [24, 25], we conducted
survival studies with heterozygous Trx1KO and heterozygous Trx2KO mice. Since Trx plays important roles in
maintaining a reduced state in the cells, we anticipated
that these mice might have higher oxidative stress/damage
and/or mitochondrial functional changes, which may lead
to a shorter lifespan.
Trx1 heterozygous knockout mice were generated with an
ES clone purchased from Lexicon Pharmaceuticals, Inc.
(OST452454), in which Trx1 expression was abolished
by gene-trap insertion into the first intron of the Trx1
gene. The young (4-6 months) and old (18-20 months)
heterozygous Trx1KO showed significantly lower (approximately 40% to 60%) Trx1 levels in the tissues examined compared to WT mice. Trx1 down-regulation did not
affect levels and activities of other antioxidant enzymes.
Trx1KO mice showed significantly lower levels of TrxASK1 complex and higher levels of ASK1 phosphorylation, which indicates that apoptosis is enhanced through
the ASK pathway. As a consequence, a slight reduction
in tumors was observed in Trx1KO compared to WT
mice at 22-24 months of age. However, Trx1KO mice
did not have remarkable changes in the survival curve
compared to WT control mice, i.e., no signifcant changes
were observed in median (50% survival) and maximum
(10% survival) lifespans compared to WT mice (Figure
2).Therefore, a reduction in Trx1 levels does not seem
to have signifcant effects on aging, although there was a
subtle indication it may attenuate age-related tumor development.
To test the effects of Trx down-regulation in the mitochondria on aging, we conducted a survival study using Trx2KO
mice [26]. Trx2KO mice were generated by gene trapping
using random insertional mutagenesis [27]. Our previous study showed that levels of Trx2 were approximately
50% less in Trx2KO mouse tissues compared to WT littermates [26]. Trx2 down-regulation did not change other
antioxidant enzymes levels and activities. Reduced Trx in
mitochondria showed that mitochondrial ATP production
and ETC activity were signifcantly reduced and ROS production and oxidative damage were increased in Trx2KO
mice. In addition, the mitochondrial apoptosis pathway was
enhanced in Trx2KO mice, resulting in higher sensitivity
to apoptosis induction in isolated cells from Trx2KO mice
[26]. Although reduced Trx in mitochondria led to mitochondrial function impairment, increased ROS production
and oxidative damage, and enhanced apoptosis in Trx2KO
mice, the survival curves of Trx2 mice showed no signifcant changes compared to WT littermates [26, 28].
Thus, in spite of several pathophysiological changes, reduced Trx in either cytosol or mitochondria alone did not
have a signifcant impact on the longevity of Trx1KO or
Trx2KO mice.
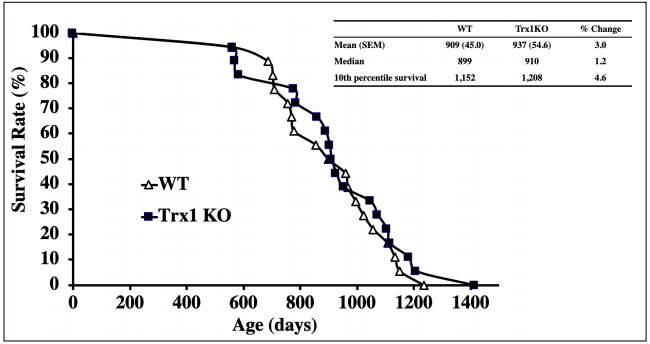
Figure 2. Survival curves of Trx1KO and WT mice. The survival curves of Trx1KO (closed squares) and WT (open triangles) mice are presented. The survival study was conducted with 18 Trx1KO mice and 18 WT male mice. No signifcant difference was observed between Trx1KO and WT mice.
Combined effects of overexpressing Trx1 and Trx2
Based on our survival studies, overexpression of Trx1 or
Trx2 alone did not show a signifcant impact on longevity
(i.e., extending the lifespan only in the earlier part of life
by Trx1 or slight extension of lifespan by Trx2), despite
interesting cellular and physiological changes. These
results indicate that Trx overexpression in only one compartment of the cell is not enough to extend the lifespan.
In other words, Trx overexpression in both the cytosol and
mitochondria may be required to have a notable impact
on aging, i.e., signifcant life extension and reducing agerelated pathology including cancer.
Therefore, we examined the effects of overexpression of
both Trx1 and Trx2 on lifespan and age-related pathology
(especially cancer) with mice overexpressing Trx in both
the cytosol and mitochondria (TXNTg x TXN2Tg) [29].
Our study showed that TXNTg x TXN2Tg mice overexpressed Trx1 (2 to 14-fold increase) and Trx2 (1.8 to
2.3-fold increase) in all tissues over the lifespan without
changes in other antioxidant enzymes levels and activities
or developmental abnormalities [29]. Contrary to our expectations, male TXNTg x TXN2Tg mice had signifcantly shorter lifespans compared to WT littermates, i.e. approximately 14.6% (mean), 16.3% (median), and 7% (10th
percentile) shorter lifespans were observed in TXNTg x
TXN2Tg compared to WT mice [29]. The cross-sectional
pathology demonstrated that TXNTg x TXN2Tg mice
had slightly higher incidences of total neoplastic diseases
and lymphoma than WT mice. TXNTg x TXN2Tg mice
showed significantly higher grades of lymphoma compared to WT mice, which indicates that overexpression of
Trx in both the cytosol and mitochondria accelerates lymphoma development during aging [29].
The shorter lifespan and accelerated tumor development
in TXNTg x TXN2Tg mice were accompanied with
changes in several redox-sensitive signaling pathways
that could play important roles in: 1) cancer growth (HIF-
1α levels and NFκB activity increase) [30-33] and 2)
apoptosis suppression and cancer development (reduced
ASK1 activity) [17]. These results were surprising since
our studies with mice overexpressing Trx in the cytosol
(Trx1) or mitochondria (Trx2) alone showed protection of
cells/tissues against oxidative stress, and no deleterious
effects were observed during aging in transgenic mice upregulating Trx1 or Trx2 [15, 16, 23]. Thus, to our knowledge, our study with TXNTg x TXN2Tg mice was the
frst report demonstrating the deleterious effects on aging
by synergetic overexpression of Trx in both the cytosol
and mitochondria.
Combined effects of down-regulatng Trx1 and Trx2
Because of the shortened lifespan and enhanced tumor
development observed in TXNTg x TXN2Tg mice, we
subsequently tested whether down-regulation of Trx in
both the cytosol and mitochondria affects aging and agerelated diseases in mice. Interestingly, survival studies using Trx1KO x Trx2KO mice showed opposite results from
TXNTg x TXN2Tg mice: an approximately 10% and 9%
extension of median lifespan in male and female Trx1KO
x Trx2KO mice compared to WT littermates, respectively.
The detailed cross-sectional pathological analysis is ongoing, and we are currently examining several signaling
pathways that play important roles in aging and cancer.
Based on our preliminary data, the severity of lymphoma
and disease burden (total number of histological changes/
body) seem to be lower in Trx1KO x Trx2KO mice
compared to WT mice. Since other antioxidant enzymes
levels/activities were similar between Trx1KO x Trx2KO
and WT mice, these data suggest that synergetic downregulation of Trx1 and Trx2 had unexpected beneficial
effects on aging. This observation is similar to the results
reported with naked mole-rats and GPx4+/- mice [34, 35].
The slight extension (9-10%) of lifespan and attenuated
tumor development in Trx1KO x Trx2KO mice were accompanied with several signaling/molecular changes: 1)
enhanced ASK1 phosphorylation; 2) increased both caspase activity and cytochrome c (cyt c) release; 3) reduced
mTOR and HIF-1α; and 4) enhanced autophagy compared
to WT mice. All of these pathways play important roles in
cell death and growth, especially in cancer.
Based on our ongoing work and preliminary observations,
we predict that reduced Trx levels in both the cytosol and
mitochondria may reduce/retard age-related cancer development through: 1) enhanced removal of damaged cells
(apoptosis) through mitochondrial and ASK1 pathways; and 2) enhanced removal of cellular damage (autophagy).
Both of these processes could be major contributing factors in reducing age-related genomic instability and attenuating age-related cancer development in Trx1KO x
Trx2KO mice. We also predict that reduced cancer development could further extend healthspan and lifespan in
Trx1KO x Trx2KO mice. We are currently seeking the underlying mechanisms of the anti-cancer effects of reduced
Trx in both the cytosol and mitochondria using a unique
mouse model.
What have we learned from the survival studies with mice overexpressing or down-regulating Trx1 and/or Trx2, and where do we go from here?
Although the pathophysiological roles of Trx and thioredoxin interacting protein (Txnip) in mammals have been
demonstrated [1, 36], the systemic examination of the effects of Trx on mammalian aging had not been achieved
until our laboratory conducted the frst systematic survival
studies with mice overexpressing or down-regulating Trx1
and Trx2. Based on our data, several interesting features
of thioredoxin and aging have been revealed: 1) overexpression of Trx1 alone [Tg(act-TRX1)+/0 and Tg(TXN)+/0
mice] extended lifespan only in the first half of life; 2)
overexpression of Trx2 alone [Tg(TXN2)+/0 mice] showed
a slight extension of lifespan; 3) Trx1 or Trx2 downregulation in Trx1KO or Trx2KO mice had little effect on
lifespan; 4) synergetic overexpression of Trx1 and Trx2
(TXNTg x TXN2Tg mice) resulted in a shorter lifespan
with accelerated tumor development; and 5) an ongoing
study using mice down-regulating Trx1 and Trx2 (Trx1KO
x Trx2KO) slightly extended lifespan and suppressed tumor development in both males and females. These results
clearly indicate that synergetic changes (overexpression
or down-regulation) in Trx1 and Trx2 are required to have
signifcant impacts on longevity, although overexpression
or down-regulation of Trx1 or Trx2 alone showed various
molecular and cellular changes.
Considering the life-extending effects of overexpressing
Trx1 or Trx2 alone (only in the earlier part of lifespan
by Tg(TXN)+/0 mice or slight extension of lifespan by
Tg(TXN2)+/0 mice), the shorter lifespan and accelerated
tumor development observed in TXNTg x TXN2Tg mice
were unexpected results. TXNTg x TXN2Tg mice showed
several signaling/molecular changes, i.e., lower phosphorylated ASK1, and increased succinate, HIF-1α, and NF-
κB p65 levels compared to WT mice, which could play
important roles in promoting age-related cancer development, possibly leading to a shorter lifespan [29].
These unexpected but very intriguing results led us to
question whether synergetic down-regulation of Trx1 and
Trx2 affects aging and age-related pathology in mice.
Trx1KO x Trx2KO mice showed an approximately 10%
and 9% extension of median lifespan in male and female
mice, respectively, compared to WT littermates. The preliminary cross-sectional pathological analysis showed that
the severity of lymphoma and disease burden were signifcantly lower in Trx1KO x Trx2KO mice compared to WT
mice. These data indicate that down-regulation of Trx in
both the cytosol and mitochondria has anti-cancer effects,
which could be one of the major contributing factors to
slight lifespan extension, although these mice could have
a potential increase in oxidative stress. These unexpected
and opposite results observed in both TXNTg x TXN2Tg
and Trx1KO x Trx2KO mice led us to the following question: why did Trx down-regulation in both the cytosol and
mitochondria reduce cancer and have a slight extension of
lifespan? We are currently conducting experiments to seek
the underlying mechanisms that reduce genomic instability and tumor development by enhanced apoptosis and
autophagy as consequences of the changes in signaling
pathways by reduced Trx1 and Trx2.
The results of these studies, including the ongoing study
with Trx1KO x Trx2KO mice: 1) provide a major advance
in our understanding and new aspect of oxidative stress
and redox regulation on cancer and aging; 2) lead us to
identify the key pathways (i.e., apoptosis and autophagy)
that prevent accumulation of damaged cells/cellular damage and genomic instability, leading to reduced cancer
formation; and 3) identify potential pharmacological
interventions (inhibition of Trx and/or downstream signaling pathways) for new prevention and/or therapy targets
to attenuate age-related cancer development and extend
healthspan.
Declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Acknowledgements
We acknowledge the Pathology Core
in the San Antonio Nathan Shock Center (P30-AG013319)
for pathological analyses.
This research was supported by the VA Merit Review
grant from the Department of Veteran Affairs (Y.I.), NIH
grant AG13319 (Y.I.), The American Federation for Aging
Research (AFAR) grant (Y.I.), and a grant from the Glenn
Foundation (Y.I.).
References
1. Arnér E S J, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. European Journal of Biochemistry, 2000, 267(20): 6102-6109.
2. Brot N, Weissbach L, Werth J, et al. Enzymatic reduction of protein-bound methionine sulfoxide. Proceedings of the National Academy of Sciences, 1981, 78(4): 2155- 2158.
3. Brot N, Weissbach H. Peptide methionine sulfoxide reductase: biochemistry and physiological role. Peptide Science, 2000, 55(4): 288-296.
4. Chae H Z, Kang S W, Rhee S G. Isoforms of mammalian peroxiredoxin that reduce peroxides in presence of thioredoxin. Methods in enzymology. Academic Press, 1999, 300: 219-226.
5. Chae H Z, Kim H J, Kang S W, et al. Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes research and clinical practice, 1999, 45(2-3): 101-112.
6. Kim K, Kim I H, Lee K Y, et al. The isolation and purification of a specific” protector” protein which inhibits enzyme inactivation by a thiol/Fe (III)/O2 mixed-function oxidation system. Journal of Biological Chemistry, 1988, 263(10): 4704-4711.
7. Levine R L, Berlett B S, Moskovitz J, et al. Methionine residues may protect proteins from critical oxidative damage. Mechanisms of ageing and development, 1999, 107(3): 323-332.
8. Tagaya Y, Maeda Y, Mitsui A, et al. ATL-derived factor (ADF), an IL-2 receptor/Tac inducer homologous to thioredoxin; possible involvement of dithiol-reduction in the IL-2 receptor induction. The EMBO journal, 1989, 8(3): 757-764.
9. Spyrou G, Enmark E, Miranda-Vizuete A, et al. Cloning and expression of a novel mammalian thioredoxin. Journal of Biological Chemistry, 1997, 272(5): 2936-2941.
10. Miranda-Vizuete A, González J C F, Gahmon G, et al. Lifespan decrease in a Caenorhabditis elegans mutant lacking TRX-1, a thioredoxin expressed in ASJ sensory neurons. FEBS letters, 2006, 580(2): 484-490.
11. Svensson M J, Larsson J. Thioredoxin-2 affects lifespan and oxidative stress in Drosophila. Hereditas, 2007, 144(1): 25-32.
12. Takagi Y, Mitsui A, Nishiyama A, et al. Overexpression of thioredoxin in transgenic mice attenuates focal ischemic brain damage. Proceedings of the National Academy of Sciences, 1999, 96(7): 4131-4136.
13. Mitsui A, Hamuro J, Nakamura H, et al. Overexpression of human thioredoxin in transgenic mice controls oxidative stress and life span. Antioxidants and Redox Signaling, 2002, 4(4): 693-696.
14. Nakamura H, Mitsui A, Yodoi J. Thioredoxin overexpression in transgenic mice. Methods in enzymology, 2002, 347: 436-440.
15. Pérez V I, Cortez L A, Lew C M, et al. Thioredoxin 1 overexpression extends mainly the earlier part of life span in mice. Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences, 2011, 66(12): 1286-1299.
16. Flores L C, Roman M G, Cunningham G M, et al. Continuous overexpression of thioredoxin 1 enhances cancer development and does not extend maximum lifespan in male C57BL/6 mice. Pathobiology of Aging & AgeRelated Diseases, 2018, 8(1): 1533754.
17. Saitoh M, Nishitoh H, Fujii M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. The EMBO journal, 1998, 17(9): 2596- 2606.
18. Hsieh C C, Papaconstantinou J. Thioredoxin-ASK1 complex levels regulate ROS-mediated p38 MAPK pathway activity in livers of aged and long-lived Snell dwarf mice. The FASEB Journal, 2006, 20(2): 259-268.
19. Powis G, Mustacich D, Coon A. The role of the redox protein thioredoxin in cell growth and cancer. Free Radical Biology and Medicine, 2000, 29(3-4): 312-322.
20. Schriner S E, Linford N J, Martin G M, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. science, 2005, 308(5730): 1909-1911.
21. Widder J D, Fraccarollo D, Galuppo P, et al. Attenuation of angiotensin II–induced vascular dysfunction and hypertension by overexpression of thioredoxin 2. Hypertension, 2009, 54(2): 338-344.
22. Zhang H, Luo Y, Zhang W, et al. Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. The American journal of pathology, 2007, 170(3): 1108- 1120.
23. Roman M G, Flores L C, Cunningham G M, et al. Thioredoxin overexpression in mitochondria showed minimum effects on aging and age-related diseases in male C57BL/6 mice. Aging Pathobiology and Therapeutics, 2020, 2(1): 20-31.
24. Matsui M, Oshima M, Oshima H, et al. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Developmental biology, 1996, 178(1): 179-185.
25. Nonn L, Williams R R, Erickson R P, et al. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Molecular and cellular biology, 2003, 23(3): 916-922.
26. Pérez V I, Lew C M, Cortez L A, et al. Thioredoxin 2 haploinsufficiency in mice results in impaired mitochondrial function and increased oxidative stress. Free Radical Biology and Medicine, 2008, 44(5): 882-892.
27. Zambrowicz B P, Friedrich G A, Buxton E C, et al. Disruption and sequence identification of 2,000 genes in mouse embryonic stem cells. Nature, 1998, 392(6676): 608- 611.
28. Pérez V I, Bokov A, Van Remmen H, et al. Is the oxidative stress theory of aging dead?. Biochimica et Biophysica Acta (BBA)-General Subjects, 2009, 1790(10): 1005- 1014.
29. Cunningham G M, Flores L C, Roman M G, et al. Thioredoxin overexpression in both the cytosol and mitochondria accelerates age-related disease and shortens lifespan in male C57BL/6 mice. Geroscience, 2018, 40(5-6): 453-468.
30. Hoesel B, Schmid J A. The complexity of NF-κB signaling in inflammation and cancer. Molecular cancer, 2013, 12(1): 1-15.
31. Huber M A, Azoitei N, Baumann B, et al. NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. The Journal of clinical investigation, 2004, 114(4): 569-581.
32. Semenza G L. Targeting HIF-1 for cancer therapy. Nature reviews cancer, 2003, 3(10): 721-732.
33. Xia Y, Shen S, Verma I M. NF-κB, an active player in human cancers. Cancer immunology research, 2014, 2(9): 823-830.
34. Andziak B, O’Connor T P, Qi W, et al. High oxidative damage levels in the longest-living rodent, the naked molerat. Aging cell, 2006, 5(6): 463-471.
35. Ran Q, Liang H, Ikeno Y, et al. Reduction in glutathione peroxidase 4 increases life span through increased sensitivity to apoptosis. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences, 2007, 62(9): 932-942.
36. Yoshihara E, Masaki S, Matsuo Y, et al. Thioredoxin/ Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Frontiers in immunology, 2014, 4: 514.